





Cập nhật về suy thận cấp: định nghĩa, chẩn đoán, sinh bệnh học và điều trị
Robert W. Schrier , Wei Wang , Brian Poole và Amit Mitra
Hà Hoàng Kiệm, BV103, HVQY, dịch từ “Acute renal failure: definitions, diagnosis, pathogenesis, and therapy”. J Clin Invest. 2004 Jul 1; 114(1): 5–14.
Tóm tắt
Suy thận cấp (ARF), đặc trưng bởi sự mất đột ngột khả năng bài tiết chất thải, khả năng cô đặc nước tiểu, bảo tồn chất điện giải và duy trì cân bằng chất lỏng của thận, là một vấn đề lâm sàng thường gặp, đặc biệt là trong đơn vị chăm sóc đặc biệt, nơi nó liên quan đến tỷ lệ tử vong từ 50% đến 80%. Trong tổng quan này, dịch tễ học và sinh lý bệnh của suy thận cấp được thảo luận, bao gồm các rối loạn mạch máu, ống thận và viêm. Đánh giá lâm sàng của suy thận cấp và ý nghĩa đối với các liệu pháp tiềm năng trong tương lai để giảm tỷ lệ tử vong cao được mô tả.
Trong những vụ đánh bom ở London trong chiến tranh thế giới lần thứ II, Bywaters và Beall đã mô tả sự mất chức năng thận cấp tính xảy ra ở những nạn nhân bị thương nặng [1]. Hoại tử ống cấp tính (ATN: acute tubular necroses) là thuật ngữ được đặt ra để mô tả thực thể lâm sàng này, bởi vì bằng chứng mô bệnh học cho thấy hoại tử loang lổ ở ống thận khi khám nghiệm tử thi. Trong lâm sàng, thuật ngữ hoại tử ống thận cấp và suy thận cấp (ARF: acute renal failure) thường được sử dụng thay thế cho nhau. Tuy nhiên, với mục đích của bài viết này, thuật ngữ suy thận cấp được chúng tôi sử dụng, chứ không phải là hoại tử ống thận cấp. Suy thận cấp được đề cập trong bài viết này sẽ không bao gồm tăng urê máu do co mạch thận có thể đảo ngược được (azotemia prerenal: Tăng nitơ máu trước thận) hoặc tắc nghẽn đường tiết niệu (azotemia postrenal: tăng nitơ máu sau thận).
Tỷ lệ tử vong của suy thận cấp là 100% trong chiến tranh thế giới lần thứ II, do sự phát triển của phương pháp thận nhân tạo cấp tính chưa được sử dụng trong lâm sàng vào thời gian này. Phương pháp thận nhân tạo cấp tính lần đầu tiên được sử dụng trong lâm sàng là ở Chiến tranh Triều Tiên năm 1950 để điều trị cho thương binh trong quân đội, điều này đã dẫn đến giảm tỷ lệ tử vong của hội chứng suy thận cấp từ khoảng 90% xuống còn khoảng 50% [2][3]. Trong nửa thế kỷ đã trôi qua, người ta đã biết nhiều về cơ chế bệnh sinh của suy thận cấp thiếu máu cục bộ và suy thận cấp trong các mô hình thực nghiệm, nhưng có rất ít sự cải thiện về tỷ lệ tử vong. Điều này có thể được giải thích: tuổi của bệnh nhân mắc suy thận cấp tiếp tục tăng và các bệnh kèm theo ngày càng phổ biến trong dân số này. Cả hai yếu tố có thể che khuất bất kỳ sự gia tăng tỉ lệ sống sót nào liên quan đến việc chăm sóc quan trọng được cải thiện.
Tuy nhiên, việc kiểm tra tỷ lệ mắc suy thận cấp trong một số cuộc xung đột quân sự có mang lại sự lạc quan [4]. Tỷ lệ mắc suy thận cấp trong các tổn thương nghiêm trọng đã giảm từ giữa chiến tranh thế giới lần thứ II và Chiến tranh Triều Tiên, và một lần nữa giữa cuộc chiến đó là cuộc chiến tranh ở Việt Nam, mặc dù không có sự khác biệt rõ ràng nào về mức độ nghiêm trọng của các thương tích. Điều khác biệt là sự hồi sức nhanh chóng đối với bệnh nhân. Bồi phụ đầy đủ dịch trên chiến trường với việc vận chuyển nhanh chóng thương binh đến bệnh viện bằng trực thăng bắt đầu trong chiến tranh Triều Tiên và được tối ưu hóa hơn nữa trong chiến tranh ở Việt Nam. Đối với những người bị thương nặng, tỷ lệ mắc suy thận cấp thiếu máu cục bộ là 1/200 trong Chiến tranh Triều Tiên và 1/600 trong chiến tranh Việt Nam [5]. Chuỗi sự kiện lịch sử này cho thấy rằng sự can thiệp sớm có thể ngăn chặn sự xuất hiện của suy thận cấp, ít nhất là trong thương binh của quân đội. Trong các nghiên cứu thực nghiệm đã chỉ ra rằng sự tiến triển từ trạng thái tăng nitơ máu liên quan đến co mạch thận trong khi chức năng ống thận còn nguyên vẹn (tăng nitơ máu trước thận) thành suy thận cấp với rối loạn chức năng ống thận xảy ra nếu tình trạng thiếu máu cục bộ thận kéo dài [6]. Hơn nữa, can thiệp sớm bằng hồi sức và truyền dịch đã được chứng minh là ngăn chặn được sự tiến triển đến tăng nitơ máu thành suy thận cấp.
Chẩn đoán suy thận cấp
Từ các nhận định trên, một câu hỏi quan trọng là làm thế nào để đảm bảo rằng việc chẩn đoán sớm bệnh co mạch thận cấp trước (suy thận cấp trước thận: ND) khi xảy ra rối loạn chức năng ống thận, điều đó giúp tăng khả năng ngăn ngừa tiến triển thành suy thận cấp. Vấn đề này, các chẩn đoán trong quá khứ dựa vào quan sát phản ứng của bệnh nhân đối với bù dịch: giảm nồng độ nitơ urê máu (BUN) là biểu hiện sự có mặt của thuốc co mạch và có thể làm đảo ngược được, trong khi tích lũy không kiểm soát được các sản phẩm nitơ cần đào thải, tức là BUN và creatinine huyết thanh, chỉ ra sự hình thành suy thận cấp. Tuy nhiên, cách tiếp cận này thường dẫn đến tình trạng quá tải lượng lớn dịch ở bệnh nhân suy thận cấp với tắc nghẽn phổi, thiếu oxy và cần điều trị sớm để hỗ trợ thở máy và/hoặc điều trị bằng thận nhân tạo. Trên nền tảng này, trọng tâm chú ý đã chuyển sang đánh giá cặn nước tiểu và sinh hóa nước tiểu để phân biệt giữa co mạch thận với chức năng ống còn nguyên vẹn và suy thận cấp đã hình thành [7]. Nếu chức năng của ống thận còn nguyên vẹn, co mạch thận có liên quan đến sự tái hấp thu natri ở ống thận được tăng cường. Cụ thể, tỷ lệ natri được lọc qua cầu thận được tái hấp thu nhanh chóng bởi các ống thận còn bình thường ở thận bị co mạch là lớn hơn 99%. Do đó, khi chất thải chứa nitơ, như creatinine và urê, tích tụ trong máu do giảm mức lọc cầu thận (GFR) thứ phát do co mạch thận với chức năng ống thận còn nguyên vẹn, sự bài tiết một phần của natri được lọc được xác định bằng phân số thải natri ký hiệu là EFNa (FE Na = [(natri nước tiểu) × (creatinine huyết tương)] / [(natri huyết tương) × (creatinine nước tiểu)]) nhỏ hơn 1%. Một ngoại lệ đối với phản ứng sinh lý này của thận bình thường trong co mạch thận là khi bệnh nhân đang được dùng thuốc lợi tiểu, bao gồm mannitol, hoặc có glucose niệu, làm giảm tái hấp thu natri ở ống và làm tăng FE Na. Gần đây, người ta đã chứng minh rằng khi có sử dụng thuốc lợi tiểu thì tỷ lệ phân số thải ure (FE urê ) dưới 35% cho thấy chức năng của ống thận còn nguyên vẹn, do đó cho phép chẩn đoán co mạch thận thay cho suy thận cấp là nguyên nhân gây ra tăng nitơ máu [8]. Ngoài ra, co mạch thận ở bệnh nhân suy thận mạn tiến triển có thể không thấy FE Na dưới 1 do sự thích nghi mạn tính với tình trạng tăng mức lọc của các nephron riêng lẻ. Cụ thể, sự giảm thích nghi trong tái hấp thu ở ống thận để duy trì cân bằng natri trong bệnh thận mạn tính có thể làm cho FE Na cao trên 1, vì vậy chẩn đoán co mạch thận trở nên khó khăn trong bối cảnh này.
Độ đặc hiệu của FE Na trong chẩn đoán phân biệt suy thận cấp chức năng và suy thận cấp thực thể khoảng 80%. Khi suy thận cấp được hình thành, khả năng cô đặc nước tiểu bị giảm; do đó, đo độ thẩm thấu nước tiểu có thể bổ sung thêm cho FE Na trong chẩn đoán phân biệt ở bệnh nhân có tăng nồng độ ure và creatinine huyết thanh. Vì tuổi cao và nồng độ protein máu thấp có thể làm giảm độ thẩm thấu nước tiểu tối đa, thông số chẩn đoán này có thể kém nhạy hơn FE Na ở bệnh nhân có tăng nitơ máu. Tăng bài tiết của các tế bào biểu mô ống thận, được xác định bằng cách kiểm tra cặn nước tiểu, đặc điểm của hình thành suy thận cấp là có trụ nước tiểu màu nâu bẩn hoặc có tế bào ống thận thoái hóa trong nước tiểu, nhưng cũng có những hạn chế trong giá trị chẩn đoán đặc biệt là suy thận cấp không vô niệu. Bảng 1 hướng dẫn cho các chỉ số tiết niệu, theo đó suy thận cấp thực thể có thể được phân biệt với suy thận cấp chức năng. Cũng cần chỉ ra rằng một số nguyên nhân gây ra suy thận cấp, bao gồm cả do thuốc cản quang và myoglobin niệu, có thể liên quan đến FE Na dưới 1 [9]. Điều này có thể liên quan đến sự hiện diện sớm của co mạch thận nặng và chức năng ống lượn xa nguyên vẹn, có thể xảy ra khi có tổn thương ống lượn gần.
Bảng 1. Hướng dẫn các chỉ số tiết niệu, theo đó suy thận cấp thực thể có thể được phân biệt với suy thận cấp chức năng khi ống thận còn nguyên vẹn (tăng nitơ máu trước thận).
|
Xét nghiệm |
Suy thận cấp chức năng |
Suy thận cấp thực thể |
|
Độ thẩm thấu nước tiểu (mOsm/kg) |
>500 |
<400 |
|
Nồng độ Na nước tiểu (mEq/l) |
<20 |
>40 |
|
Tỉ số Ure máu/creatinin máu |
>40 |
<20 |
|
Phân số thải Na: EFNa (%) |
<1 |
>2 |
|
Phân số thải ure: EFUre (%) |
<35 |
>35 |
|
Cặn nước tiểu |
Bình thường; Có thể có trụ hyalin hoặc trụ hạt mịn |
Có tế bào biểu mô ống thận; Trụ hạt hoặc trụ màu nâu bẩn |
Các kết quả quan sát gần đây cho thấy đối với những bệnh nhân có nồng độ BUN tăng và/hoặc nồng độ creatinine huyết thanh tăng, tham khảo ý kiến sớm với bác sĩ chuyên khoa thận có thể làm giảm sự hình thành và giảm tỉ lệ tử vong của suy thận cấp [10]. Lý do giải thích cho kết quả quan sát này không rõ ràng nhưng có thể liên quan đến việc đánh giá sớm bởi bác sĩ thận không chỉ căn cứ vào xét nghiệm máu mà còn cả các xét nghiệm sinh hóa nước tiểu, giảm tỷ lệ quá tải dịch và cần thở máy, và có thể còn do bắt đầu lọc máu sớm hơn. Trên nền tảng này, việc tìm kiếm các thông số nhạy cảm để chẩn đoán sớm suy thận cấp và định nghĩa được chấp nhận thống nhất về suy thận cấp đã được đề xuất. Một định nghĩa lâm sàng về suy thận cấp được tìm kiếm tập trung vào mức độ tích lũy chất thải nitơ trong máu, như creatinine huyết thanh, BUN, hoặc cystatin C [11] để chẩn đoán sớm. Cách tiếp cận này, tuy nhiên, có những hạn chế quan trọng. Với suy thận cấp đã hình thành, mức lọc cầu thận thường dưới 10 ml/phút. Lượng nitơ tích lũy trong máu phụ thuộc vào khoảng thời gian bệnh nhân có mức lọc cầu thận dưới 10 ml/phút. Do đó, một bệnh nhân có tình trạng tăng dị hóa với suy thận cấp đã hình thành có thể có nồng độ creatinine huyết thanh là 1,8 mg/dl trong vòng 24 giờ đầu tiên của suy thận cấp, nhưng cũng có một số bệnh nhân có thể có nồng độ 1,0 mg/dl sau 5 ngày kể từ khi xảy ra suy thận cấp, mặc dù mức lọc cầu thận đã ít hơn 10 ml/phút trong cả hai trường hợp. Tuy nhiên, việc chấp nhận tăng creatinine huyết thanh cấp tính 50% trở lên với nồng độ trên 2,0 mg/dl trong lâm sàng được xác định là suy thận cấp có thể làm tăng khả năng can thiệp sớm và mang lại sự nhất quán hơn cho các nhóm bệnh nhân trong các thử nghiệm can thiệp trong tương lai. Tuy nhiên, định nghĩa như vậy không phân biệt được giữa tăng creatinine trong huyết thanh là hậu quả của (a) tình trạng tăng nitơ phi protein do mạch máu thận với tăng nitơ phi protein máu có tổn thương chức năng ống thận, chẳng hạn như xảy ra với giảm thể tích do suy tim hoặc suy gan tiến triển; (b) tình trạng tăng nitơ phi protein máu sau thận do tắc nghẽn đường tiết niệu; hoặc (c) suy thận cấp do thiếu máu cục bộ cấp tính và/hoặc nhiễm độc thận. Một cách tiếp cận nhạy cảm để loại trừ tăng nitơ phi protein máu sau thận là xác định lượng nước tiểu trong bàng quang còn sót lại dưới 50 ml và loại trừ giãn đài bể thận bằng siêu âm thận.
Tuy nhiên, sự khác biệt của tăng nitơ phi protein máu do suy thận cấp chức năng và suy thận cấp thực thể là khó khăn. Một số dấu ấn sinh học đã được đề xuất để chẩn đoán sớm suy thận cấp và hiện đang được nghiên cứu. Chúng bao gồm tăng bài tiết qua nước tiểu của phân tử tổn thương thận-1 [12], IL-18 [13] và enzyme ống thận [14]. Để bất kỳ thông số nào trong các thông số này hoặc các dấu ấn sinh học tiềm năng khác trong tương lai trở nên thiết thực trong chẩn đoán lâm sàng sớm suy thận cấp, phải xem xét tính đặc hiệu, độ nhạy, tính tiện lợi, hiệu quả chi phí và lợi thế so với phương pháp sử dụng FE Na rẻ tiền và sẵn có.
Từ quan điểm chẩn đoán lâm sàng, chúng ta cần thỏa thuận sử dụng thuật ngữ suy thận cấp, chứ không phải hoại tử ống thận cấp, không phụ thuộc vào việc tổn thương thận cấp tính là do thiếu máu cục bộ hay do nhiễm độc thận. Tuy nhiên, một thỏa thuận như vậy cũng sẽ bắt buộc phải sử dụng thuật ngữ tăng nitơ phi protein máu trước thận và tăng nitơ phi protein máu sau thận cho những tình trạng có khả năng hồi phục chức năng thận do co mạch thận và tắc nghẽn đường tiết niệu, tương ứng. Ngoài ra, phải công nhận rằng chẩn đoán suy thận cấp chỉ dựa trên mức tăng creatinine huyết thanh, BUN hoặc cystatin C sẽ bao gồm nhiều bệnh nhân không mắc hội chứng suy thận cấp thực thể, một thực tế không dễ bị đảo ngược bởi bù dịch trong hồi sức để cải thiện chức năng tim hoặc gan, hoặc giảm tắc nghẽn đường tiết niệu.
Cơ chế bệnh sinh của suy thận cấp
Dựa trên các phân tích đã nói ở trên, thảo luận về cơ chế của suy thận cấp trong bài này sẽ không bao gồm tăng nitơ phi protein máu do co mạch thận với chức năng ống thận còn nguyên vẹn (tăng nitơ phi protein máu trước thận) hoặc tắc nghẽn đường tiết niệu (tăng nitơ phi protein máu sau thận). Các cơ chế của suy thận cấp liên quan đến cả các yếu tố mạch máu và ống thận [15]. Nói chung, hậu quả của thiếu máu cục bộ đối với thận sẽ là nguyên nhân của suy thận cấp cũng được thảo luận ở đây. Trong khi sự giảm lưu lượng máu thận với việc giảm oxy và các chất cần thiết cho chuyển hóa của các tế bào ống thận cũng là một yếu tố quan trọng trong thiếu máu cục bộ thận, cũng cần nhớ rằng sự gia tăng tương đối nhu cầu oxy của ống thận cũng là một yếu tố gây thiếu máu cục bộ thận.
Thuật ngữ hoại tử ống thận cấp, mặc dù thường được sử dụng để mô tả hội chứng suy thận cấp trong lâm sàng trong trường hợp không có tăng nitơ phi protein máu trước thận và sau thận, có phần sai lệch. Cụ thể, khi suy thận cấp xảy ra, sự hiện diện của hoại tử ống thận khi kiểm tra mô bệnh học của thận chỉ thỉnh thoảng mới nhìn thấy trong các tế bào ống thận và, trong một số trường hợp, thậm chí có thể không phát hiện được [16]. Tuy nhiên, điều rõ ràng với suy thận cấp đã xảy ra là các cầu thận bình thường về mặt hình thái. Điều này có lẽ biện minh cho lý do các tác giả tập trung điều tra vào ống thận. Các cuộc thảo luận sau đây sẽ mô tả một số rối loạn mạch máu, ống thận và viêm xảy ra với một nhiễm độc thận cấp tính. Một nỗ lực sau đó sẽ được thực hiện để tích hợp các yếu tố gây bệnh tiềm ẩn quan trọng.
Bất thường mạch máu thận
Mất khả năng tự tự điều chỉnh dòng máu thận và tăng co mạch thận: vai trò của tăng calci vào tế bào và ty thể.
Tổn thương thiếu máu cục bộ cấp tính đã được chứng minh ở động vật thực nghiệm có liên quan đến mất khả năng tự điều chỉnh dòng máu thận [17]. Hơn nữa, thay vì bình thường giãn mạch thận tự động xảy ra trong quá trình giảm áp lực tưới máu thận, nhưng có bằng chứng cho thấy rằng co mạch thận thực sự đã xảy ra ở thận bị thiếu máu cục bộ. Một sự gia tăng trong đáp ứng với kích thích thần kinh thận cũng đã được quan sát liên quan đến tổn thương thiếu máu cục bộ cấp tính [17]. Hơn nữa, đáp ứng với thuốc co mạch norepinephrine và endothelin ngoại sinh tăng lên đã được quan sát thấy ở thận thiếu máu cục bộ cấp tính [18]. Những bất thường mạch máu quan sát thấy ở thận thiếu máu cục bộ có thể liên quan đến kết quả gia tăng lượng calci vào tế bào được quan sát thấy trong các động mạch đến của cầu thận. Các thuốc chẹn kênh calci nội bào có thể làm đảo ngược sự mất khả năng tự điều chỉnh dòng máu thận và làm tăng độ nhạy với kích thích thần kinh thận [17], làm giảm vai trò gây bệnh của tăng nồng độ calci nội bào trong động mạch đến của cầu thận thiếu máu cục bộ. Sự tích lũy calci trong ty thể ở thận thiếu máu cục bộ cũng đã được đảo ngược bằng cách sử dụng thuốc chẹn kênh calci [19-21]. Hơn nữa, thuốc chẹn kênh calci cũng đã được chứng minh là làm giảm rối loạn chức năng thận và nhiễm độc thận liên quan đến thuốc cyclosporine sau ghép thận, khi được sử dụng trước khi dùng thuốc và thiếu máu cục bộ [22].
Xung huyết tủy ngoài
Xung huyết tủy ngoài của thận là một dấu hiệu mạch máu khác của thiếu máu cục bộ thận cấp tính. Sự xung huyết này làm xấu đi tình trạng thiếu oxy tương đối ở tủy ngoài và do đó làm tổn thương do thiếu oxy ở đoạn S3 của ống lượn gần và của quai Henle phần dày (Hình 1) [23]. Sự điều hòa của các phân tử bám dính có liên quan đến ứ huyết ở tủy ngoài. Các kháng thể kháng ICAM và P-selectin đã được chứng minh là có khả năng bảo vệ chống lại tổn thương do thiếu máu cục bộ thận cấp tính [24-27]. Thiếu máu thận cấp tính cũng đã được chứng minh là có liên quan đến tổn thương nội mô, có lẽ ít nhất một phần là do tổn thương thiếu oxy tăng. Một số bằng chứng ủng hộ khả năng này đã được quan sát là bạch cầu hoạt hóa tăng do tổn thương thiếu máu cục bộ thận, nhưng quan sát này không được ủng hộ bằng cách sử dụng bạch cầu từ bệnh nhân mắc bệnh u hạt mạn tính, các bạch cầu này không bị hoạt hóa đáp ứng với thiếu oxy [28]. Tổn thương oxy hóa cũng có thể dẫn đến giảm eNOS và prostaglandin giãn mạch cũng như tăng endothelin, tất cả đều có thể làm tăng tác dụng co mạch ở thận của các thuốc tăng áp tuần hoàn dùng trong suy thận cấp [29-31].
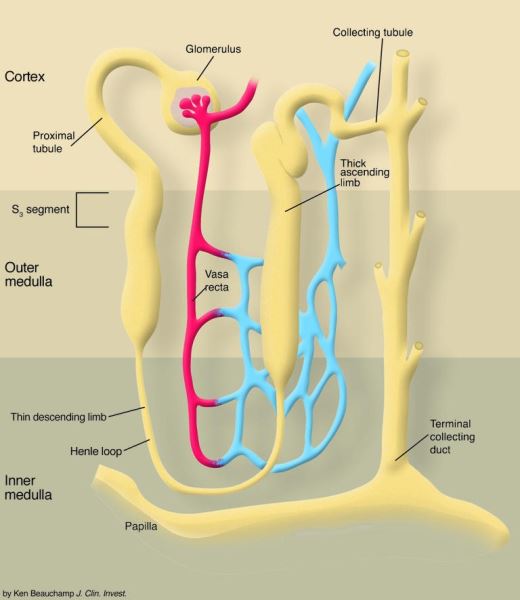
Hình 1. Tình trạng thiếu oxy tương đối ở tủy ngoài có xu hướng dẫn đến tổn thương do thiếu máu cục bộ ở đoạn S3 của ống lượn gần. Nhánh lên dày của quai Henle cũng nằm trong khu vực thiếu oxy này của thận và, tùy thuộc vào nhu cầu tái hấp thu ở ống, cũng có thể bị tổn thương do thiếu máu cục bộ. Tuy nhiên, nhánh lên dày của quai Henle có thể được bảo vệ nhiều hơn chống lại tổn thương do thiếu máu cục bộ, bởi vì phân đoạn nephron này tiêu thụ nhiều glucose để tổng hợp ATP hơn phân khúc S3 .
Với phổ các hậu quả mạch máu quan sát được liên quan đến thiếu máu cục bộ cấp tính (Hình 2), một gợi ý trước đó đã được đưa ra rằng thuật ngữ hoại tử ống thận cấp nên được thay đổi bằng thuật ngữ bệnh lý mạch máu. Tuy nhiên, thuốc giãn mạch thận, đưa lưu lượng máu thận trở lại bình thường trong các mô hình động vật suy thận cấp thực nghiệm và người có suy thận cấp được thiết lập, đã không chứng minh được là làm tăng mức lọc cầu thận. Do đó, trong những năm gần đây, việc tìm kiếm các cơ chế trung gian của suy thận cấp tập trung chủ yếu vào ống thận.
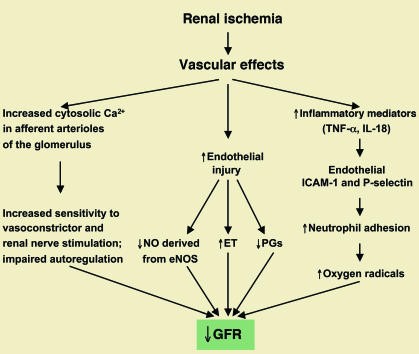
Hình 2. Các yếu tố mạch máu góp phần vào sinh bệnh học của suy thận cấp thiếu máu cục bộ. ET: endothelin; PG: prostaglandin. Hình được sửa đổi với sự cho phép của Tạp chí Thận học [15].
Bất thường ống thận
Không có rối loạn chức năng ống thận xảy ra trong suy thận cấp cho đến khi tái hấp thu natri ở ống bị giảm, tức là FE Na > 2.0, tại thời điểm ống thận bình thường, ống thận tăng khả năng tái hấp thu natri để đáp ứng với co thắt mạch máu thận. Tuy nhiên, các nghiên cứu về các bất thường ở ống thận xảy ra sau khi bị tổn thương thiếu máu cục bộ thận cấp, phải chứng minh được bằng cách nào để quan sát được rối loạn chức năng ống thận qua trung gian nào làm giảm mức lọc cầu thận xuống dưới 10% so với bình thường - đặc trưng của suy thận cấp.
Thay đổi cấu trúc trong suy thận cấp thiếu máu cục bộ
Suy thận cấp được đặc trưng bởi rối loạn chức năng ống thận với sự suy giảm tái hấp thu natri và nước và liên quan đến sự bong tróc và bài tiết của màng tế bào ống lượn gần và tế bào ống biểu mô thận vào nước tiểu [32] ( Hình 3). Khoảng 30 - 70% các tế bào có trong nước tiểu là tế bào biểu mô ống thận và có thể nuôi cấy được trong môi trường nuôi cấy [33]. Các nghiên cứu gần đây sử dụng các tế bào và kỹ thuật phân tử đã cung cấp thông tin liên quan đến các bất thường về cấu trúc của các ống thận bị tổn thương xảy ra cả trong in vitro và in vivo. Các nghiên cứu in vitro trong điều kiện thiếu oxy đã cho thấy những bất thường ở tế bào ống lượn gần có liên quan đến sự chuyển vị trí của Na+/ K+ -ATPase từ đường cơ sở sang màng đáy [34] ( Hình 3). Một so sánh giữa thận ghép được lấy từ người chết não có chức năng thận ghép chậm so sánh với thận ghép có chức năng bình thường sớm cũng cho kết quả quan trọng về vai trò của Na+/K+ -ATPase trong tổn thương thận do thiếu máu cục bộ [35]. Nghiên cứu này đã chứng minh rằng, so với thận có chức năng thận ghép bình thường sớm, những người có chức năng ghép chậm trễ có nồng độ tế bào chất của Na+/K+ -ATPase và protein gắn với actin - Spectrin (còn được gọi là fodrin) và ankyrin cao hơn đáng kể chuyển từ màng đáy sang tế bào chất (Hình 4). Sự dịch chuyển Na+/K+ -ATPase từ màng đáy sang tế bào chất có thể giải thích sự giảm tái hấp thu natri ở ống thận xảy ra ở bệnh nhân suy thận cấp. Các cơ chế theo đó vị trí quan trọng của Na+/K+ -ATPase trong màng đáy, tạo điều kiện cho việc vận chuyển natri theo vec tơ vận chuyển, không bị thiếu oxy hoặc thiếu máu cục bộ là một trọng tâm nghiên cứu. Các protein liên kết với Actin, Spectrin và ankyrin, đóng vai trò là chất nền cho calpain cysteine được hoạt hóa bằng canxi [36] (Hình 5). Về vấn đề này, các nghiên cứu in vitro ở ống lượn gần đã cho thấy có sự gia tăng nhanh chóng nồng độ canxi trong tế bào trong tình trạng thiếu oxy cấp tính, kết quả này chống lại bằng chứng tổn thương ống thận do giải phóng lactic dehydrogenase (LDH) [37] (Hình 6).
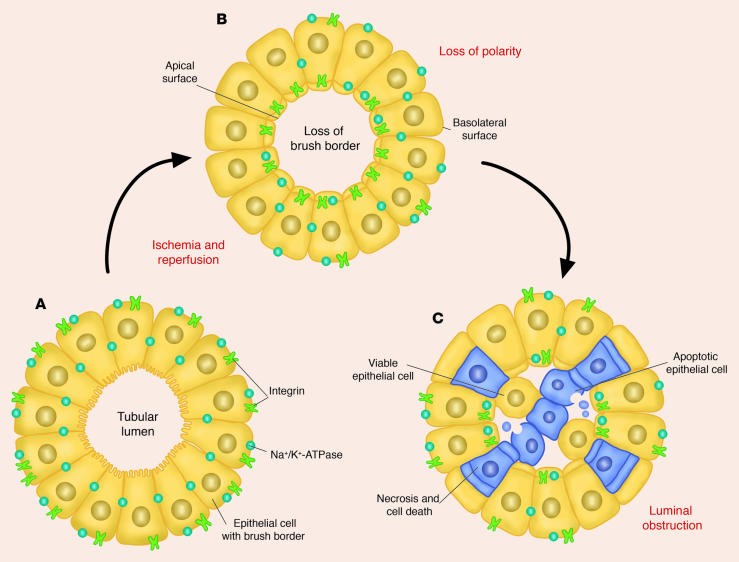
Hình 3. Sau thiếu máu cục bộ và tái tưới máu, những thay đổi hình thái xảy ra ở ống lượn gần, bao gồm mất phân cực, mất đường viền bàn chải và phân phối lại integrins và Na+/K+ -ATPase lên bề mặt đỉnh. Calci và các gốc oxy cũng có thể có vai trò trong những thay đổi hình thái này, ngoài việc chết tế bào sau đó hoại tử và chết tế bào theo chương trình. Cả hai loại tế bào còn khả năng sống và không còn khả năng sống được bong vào lòng ống thận, dẫn đến sự hình thành các trụ hình và gây tắc nghẽn ống thận, góp phần làm giảm mức lọc cầu thận. Hình được sửa đổi với sự cho phép của Tạp chí Y học New England [94].
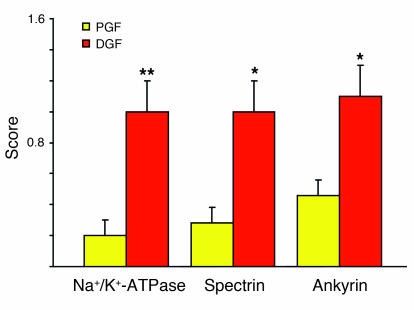
Hình 4. Nhuộm miễn dịch huỳnh quang cho thấy vị trí tế bào của các protein liên kết actin ankyrin, Spectrin và Na+/K+ -ATPase ở thận ghép lấy từ người chết não với chức năng ghép kịp thời (PGF) và chức năng ghép chậm (DGF) [35]. Các phần nhuộm màu được kiểm tra ở độ phóng đại × 60 - 600 bằng cách sử dụng kính hiển vi điện tử quét laser. Ghi điểm được thực hiện như sau: 0, thuốc nhuộm bắt màu liên tục ở màng đáy; 0,5, 1,0 và 1,5, thuốc nhuộm bắt màu không liên tục ở màng đáy tương ứng với ít hơn 50%, xấp xỉ 50% và lớn hơn 50%, và chất nhuộm màu xuất hiện trong tế bào chất. Ở thận có chức năng ghép chậm, khoảng 50% ankyrin, Spectrin và Na+/K+ -ATPase được chuyển từ màng đáy sang tế bào chất, trong khi những thận có chức năng ghép nhanh chóng chỉ có sự dịch chuyển tối thiểu các protein này từ màng đáy. * P <0,01 so với PGF, ** P <0,05 so với PGF.
.jpg)
Hình 5. Nghiên cứu hình thái tế bào của protease cystein trong tình trạng thiếu oxy/thiếu máu cục bộ ở ống lượn gần. Màng đáy của ống lượn gần chứa các tiểu đơn vị Na+/K+ -ATPase, được liên kết với Actin ở khung tế bào với ankyrin và Spectrin. Điều này tạo thành một phức hợp trao đổi chất ổn định. Hình thái tế bào của protease cystein trong tình trạng thiếu oxy/thiếu máu cục bộ được hiển thị. Ank: ankyrin; CAM: phân tử kết dính tế bào; 4.1: protein 4.1. Hình được sao chép với sự cho phép của Taylor & Francis [36].
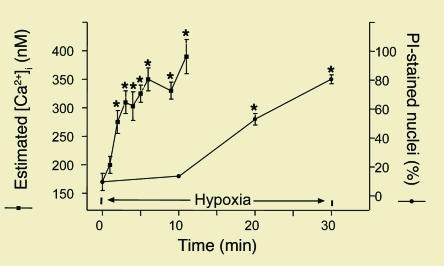
Hình 6. Tăng nồng độ Ca2+ tự do trong nội bào ống lượn gần trong tình trạng thiếu oxy (được đo bằng chỉ số Ca2+ huỳnh quang, chất chỉ thị Fura-2) trong tổn thương màng tế bào, được đánh giá bằng nhuộm propidium iodide (PI) [37]. * có ý nghĩa so với thời gian 0.
Có nhiều bằng chứng ủng hộ tầm quan trọng của việc chuyển vị trí Na+/K+ -ATPase từ màng đáy sang tế bào chất trong quá trình thiếu máu cục bộ/tái tưới máu thận. Cụ thể, các sản phẩm phân hủy qua trung gian calpain của phổ protein liên kết với Actin xảy ra với thiếu máu cục bộ thận đã được chứng minh. Hoạt động của Calpain tăng lên trong tình trạng thiếu oxy ở ống lượn gần bị cô lập cũng cũng được chứng minh [38]. Đo lường giải phóng LDH sau khi ức chế calpain đã chứng minh sự suy giảm tổn thương do thiếu oxy đối với ống lượn gần [39]. Không có bằng chứng tăng cathepsin, một loại protease cystein khác ở ống lượn gần trong tình trạng thiếu oxy. Các nghiên cứu sâu hơn đã chứng minh một con đường độc lập với calci để kích hoạt calpain trong tình trạng thiếu oxy. Calpastatin, một chất ức chế hoạt hóa calpain nội bào, đã được chứng minh là bị giảm trong quá trình thiếu oxy liên quan đến sự gia tăng của một protease cysteine khác, caspase [40]. Tác dụng này của hoạt động calpastatin giảm có thể được đảo ngược bằng ức chế caspase. Hình 7, minh họa các con đường phân giải protein có thể liên quan đến tổn thương tế bào ống thận qua trung gian calpain trong quá trình thiếu oxy. Hoạt hóa calci của phospholipase A cũng đã được chứng minh là góp phần gây tổn thương ống thận trong thiếu máu cục bộ [41].
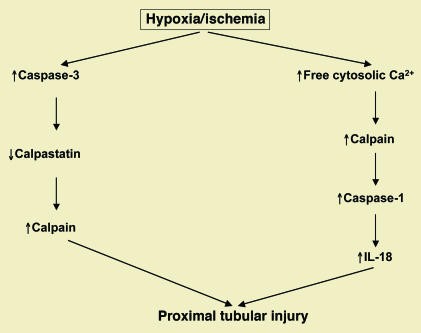
Hình 7. Thiếu oxy/hoại tử ống thận do thiếu máu cục bộ dẫn đến kích hoạt các con đường protease cysteine liên quan đến calpains và cả caspase-1 (một caspase viêm) và caspase-3 (một caspase liên quan đến chết tế bào theo chương trình). Calpain được kích hoạt cả bằng con đường tăng Ca2+ nội bào và giảm calpastatin. Calpain sau đó kích hoạt caspase-1, chất kích thích cytokine viêm IL-18. Caspase-3 cắt calpastatin [40].
Tắc nghẽn ống thận trong suy thận cấp thiếu máu cục bộ
Sự tồn tại của các con đường phân giải protein liên quan đến protease cystein, cụ thể là calpain và caspase có thể giải thích sự giảm tái hấp thu natri ở ống lượn gần và tăng FE Na thứ cấp do sự phân ly protein của Na+/K+ -ATPase từ màng đáy của tế bào. Tuy nhiên, tổn thương đơn độc ống thận không giải thích được sự sụt giảm mức lọc cầu thận dẫn đến sự tích lũy chất thải nitơ và dẫn đến sự gia tăng BUN và creatinine huyết thanh. Tuy nhiên, có những con đường tiềm năng trong đó xảy ra và không xảy ra mất màng diềm bàn chải, mất tế bào ống lượn gần, và giảm tái hấp thu natri ở ống lượn gần có thể dẫn đến giảm mức lọc cầu thận trong suy thận cấp. Trước hết, màng diềm bàn chải và các mảnh vụn của tế bào có thể cung cấp chất nền cho các trụ trong lòng ống thận gây ra sự tắc nghẽn ống thận tạo ra sức kháng cao của các nephron (Hình 3). Trên thực tế, các vi đoạn của từng nephron thận ở bệnh nhân suy thận cấp đã chứng minh các trụ hình gây tắc nghẽn trong lòng ống lượn xa và góp [42]. Quan sát này có thể giải thích các ống lượn gần bị giãn được quan sát khi sinh thiết thận ở bệnh nhân suy thận cấp, mặc dù mức lọc cầu thận giảm thấp hơn 10% so với bình thường. Các trụ trong lòng ống thận ở bệnh nhân suy thận cấp được nhuộm màu cho thấy chúng là protein Tamm-Horsfall (THP), được sản xuất ở nhánh lên dày của quai Henle (Hình 8). Điều quan trọng, protein Tamm-Horsfall được tiết vào dịch ống như một monome nhưng sau đó có thể trở thành một polymer tạo thành một chất giống như gel do tăng nồng độ Na+ ở dịch lòng ống thận, xảy ra ở ống lượn xa trong suy thận cấp mà lâm sàng là sự giảm tái hấp thu natri ở ống thận [43]. Do đó, gel polymer protein Tamm-Horsfall trong lòng ống lượn xa tạo ra một chất nền trong trong lòng ống để hình thành các thể hình trụ và có chứa bên trong các tế bào chết theo chương trình và tế bào biểu mô ống thận hoại tử, màng diềm bàn chải và ECM (các chất ngoại bào) (hình 3) [44]. Liệu tắc nghẽn ống thận do các trụ có đủ để giải thích cho việc giảm mức lọc cầu thận liên quan đến suy thận cấp lâm sàng hay không. Chắc chắn, áp lực lọc xuyên màng mao mạch cầu thận có thể giảm thứ phát do tăng áp lực trong lòng ống thận, như đã được chứng minh bằng cách sử dụng kỹ thuật vi chọc hút ở động vật thực nghiệm bị tắc nghẽn niệu quản cấp tính [45]. Tuy nhiên, các nghiên cứu vi chọc hút ở thận bị tổn thương do thiếu máu cục bộ cấp tính đã chứng minh rằng tốc độ dòng dịch trong ống thận ở một số ống thận có thể đánh bật một số trụ gây tắc nghẽn trước đó và cải thiện mức lọc cầu thận ở nephron đó [46]. Vì vậy, có thể đề xuất rằng ít nhất một số trụ sẽ không gây tắc nghẽn dòng chảy ống thận nếu áp lực cầu thận và áp lực ống thận là bình thường.
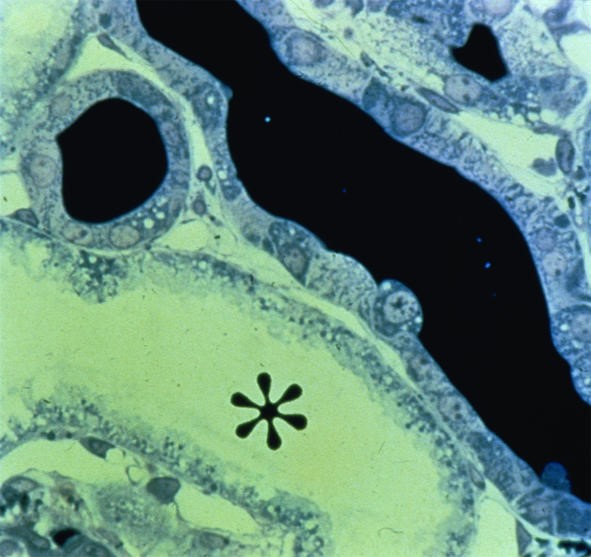
Hình 8. Nhuộm protein Tamm-Horsfall thấy trụ trong lòng ống thận (màu đen: ND) và ống thận bị giãn trong suy thận cấp thiếu máu cục bộ. Dấu hoa thị cho thấy lòng ống thận hình ống bình thường. × 100.
Có một số bằng chứng trong thực nghiệm cho thấy các tế bào biểu mô ống thận bong vào lòng ống khi thiếu máu cục bộ có thể bám vào các tế bào ống thận khác cùng với các chất ngoại bào cũng gây ra tắc nghẽn ống thận. Sự kết dính tế bào này trong thiếu máu cục bộ thận cấp đã được chứng minh có liên quan đến các phân tử bám dính qua trung gian integrin thông qua liên kết với các chuỗi Arg-Gly-Asp (RGD) [47]. Để hỗ trợ khả năng này, các hợp chất RGD tổng hợp theo chu kỳ được sử dụng trong giai đoạn tái tưới máu đã được chứng minh là làm giảm sự tắc nghẽn ống và sự đảo ngược được gia tăng áp lực trong lòng ống lượn gần [48]. Giảm sự tắc nghẽn ống thận trong suy thận cấp thực nghiệm, được đánh giá bằng phương pháp vi chọc hút nephron, cũng đã được chứng minh khi sử dụng một chất lợi tiểu hòa tan như mannitol [49].
Cân bằng cầu - ống thận và dò ngược dịch lọc trong lòng ống thận trong suy thận cấp thiếu máu cục bộ
Việc giảm tái hấp thu natri ở ống lượn gần có liên quan đến tổn thương thiếu máu cục bộ cấp tính sẽ làm tăng việc cung cấp natri clorua cho bộ máy cận cầu thận (macula densa) gây kích hoạt cơ chế phản hồi ngược cầu - ống thận và làm giảm mức lọc cầu thận [50]. Các nghiên cứu tưới máu vi chọc hút cung cấp natri clorua tăng lên cho bộ máy cận cầu thận đã chứng minh làm giảm mức lọc cầu thận của nephron tương ứng tới 50% [50]. Tuy nhiên, mức độ giảm mức lọc cầu thận này không thể giải thích được sự giảm mức lọc cầu thận lớn hơn nhiều của suy thận cấp trong lâm sàng. Tuy nhiên, do vai trò của động mạch đến ở cầu thận trong cơ chế điều chỉnh cầu - ống thận, nên tăng độ nhạy của động mạch đến của cầu thận gây co mạch mạnh, như đã thảo luận trước đó [18], có thể làm tăng độ nhạy cảm phản ứng của ống thận ở bệnh nhân bị suy thận cấp. Hơn nữa, sự kết hợp hình thành trụ trong ống thận và kích hoạt cơ chế phản hồi cầu - ống thận trong thiếu máu cục bộ thận cấp, cả hai có thể phối hợp với sự giảm hấp thu natri ở ống lượn gần, điều này có thể giải thích thỏa đáng cho sự sụt giảm nghiêm trọng mức lọc cầu thận trong suy thận cấp lâm sàng. Vai trò của phản hồi cầu - ống thận trong suy thận cấp thiếu máu cục bộ có thể là một tiềm năng có lợi cần được xem xét. Làm giảm cơ chế phản hồi cầu - ống thận để hạn chế giảm mức lọc cầu thận trong thiếu máu cục bộ thận cấp bằng cách giảm cung cấp natri clorua cho các ống thận bị tổn thương, sẽ làm giảm kích hoạt cơ chế phản hồi ngược cầu - ống thận.
Việc mất hàng rào tế bào biểu mô ống thận và/hoặc các mối nối chặt chẽ giữa các tế bào ống thận [51] trong thiếu máu cục bộ thận cấp có thể dẫn đến rò rỉ dịch lọc cầu thận trở lại hệ tuần hoàn. Nếu điều này xảy ra và thông thường các chất không thể tái hấp thu, chẳng hạn như inulin, rò rỉ trở lại tuần hoàn, thì mức lọc cầu thận thấp giả tạo sẽ xảy ra khi đo độ thanh thải inulin. Tuy nhiên, cần lưu ý rằng mức độ tổn thương ống thận lan rộng được quan sát trong các nghiên cứu thực nghiệm chứng minh tình trạng chảy ngược chất lỏng ở ống thận hiếm khi được quan sát với suy thận cấp lâm sàng ở người [52]. Hơn nữa, các nghiên cứu sàng lọc dextran ở bệnh nhân suy thận cấp đã chứng minh rằng chỉ giảm 10% mức lọc cầu thận có thể được giải thích bằng dò ngược của dịch lọc [53]. Tuy nhiên, thận ghép lấy từ người chết não có chức năng thận ghép chậm, có thể bị hoại tử ống thận nghiêm trọng, và do đó, quá trình dò ngược dịch lọc có thể là cơ chế quan trọng hơn trong trường hợp này. Các cơ chế tiềm năng khác tác động lên ống thận có thể làm giảm mức lọc cầu thận trong tổn thương thận do thiếu máu cục bộ được thể hiện trong Hình 9.
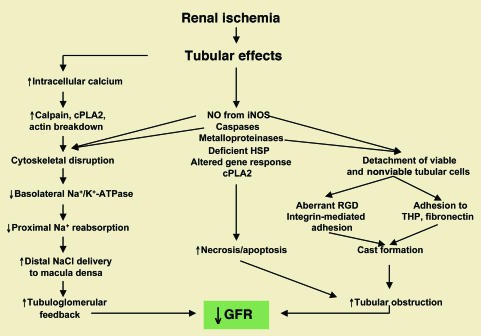
Hình 9. Ảnh hưởng của thiếu máu cục bộ lên ống thận trong cơ chế bệnh sinh của suy thận cấp thiếu máu cục bộ. cPLA2: phospholipase cytosolic A2; HSP: protein sốc nhiệt. Hình được sửa đổi với sự cho phép của Tạp chí Thận học [15].
Viêm
Hiện nay có bằng chứng đáng kể về liên quan của viêm trong cơ chế bệnh sinh của giảm mức lọc cầu thận liên quan đến tổn thương thiếu máu cục bộ thận cấp. Về vấn đề này, có bằng chứng thực nghiệm cho thấy iNOS có thể góp phần gây tổn thương ở ống thận trong suy thận cấp. Thực nghiệm gây thiếu oxy trong ống lượn gần cô lập đã chứng minh là làm tăng giải phóng NO [54] và phân tích blot Western trong homogenates thận thiếu máu cục bộ đã chứng minh có tăng protein iNOS [55]. Một kháng nguyên oligonucleotide đã cho thấy có tác dụng ngăn chặn tác dụng của iNOS và có khả năng bảo vệ chức năng ống thận chống lại tình trạng thiếu máu cục bộ thận cấp tính [55]. Hơn nữa, khi nghiên cứu iNOS, eNOS và NO trung tính tổng hợp (nNOS) trên các ống lượn gần cô lập, ở chuột phơi nhiễm với tình trạng thiếu oxy, chỉ các ống thận từ chuột bị loại ra khỏi iNOS được bảo vệ chống lại tình trạng thiếu oxy, được đánh giá bởi sự giải phóng LDH [56]. Chuột được loại iNOS cũng được chứng minh là có tỷ lệ tử vong thấp hơn khi thiếu máu cục bộ/tái tưới máu so với chuột có iNOS [57].
Cũng có bằng chứng cho thấy việc làm mất đi NO bởi các gốc oxy đã tạo ra peroxynitrite gây ra tổn thương ống thận trong thiếu máu cục bộ [58 – 60]. Việc sử dụng hormon kích thích α-melanocyte, (αMSH) có tác dụng bảo vệ chống lại tổn thương thận do thiếu máu cục bộ/tái tưới máu bằng cách ngăn chặn cả việc tạo ra iNOS và thâm nhiễm bạch cầu vào thận khi thiếu máu cục bộ [61]. Những tác nhân dọn dẹp gốc oxy tự do, như superoxide effutase, cũng đã được chứng minh là có tác dụng bảo vệ chống lại tổn thương thận cấp tính liên quan đến nhiễm độc nội độc tố [62]. Các chất ức chế Caspase, chất kháng IL-18 cũng đã được chứng minh là có tác dụng bảo vệ chống lại tổn thương thận do thiếu máu cục bộ/tái tưới máu [63, 64].
Mặc dù iNOS có thể góp phần gây tổn thương ống thận do thiếu máu cục bộ, nhưng có bằng chứng cho thấy tác dụng lên mạch máu của eNOS ở động mạch đến của cầu thận có tác dụng bảo vệ chống lại tổn thương do thiếu máu cục bộ. Về vấn đề này, ở chuột được loại bỏ eNOS đã được chứng minh là nhạy cảm hơn với tổn thương liên quan đến nội độc tố so với chuột bình thường [65]. Hơn nữa, vai trò bảo vệ của eNOS trên mạch máu có thể quan trọng hơn tác dụng xấu của iNOS ở mức độ ống thận trong quá trình thiếu máu cục bộ thận. Cơ sở cho kết luận này là quan sát việc điều trị chuột bằng chất ức chế tổng hợp NO (NOS) không đặc hiệu L -NAME, ngăn chặn cả iNOS và eNOS, làm tổn thương thiếu máu cục bộ thận so với điều trị bằng giả dược [66]. Người ta cũng chứng minh rằng NO có thể điều hòa quá mức eNOS [67] và là chất gây cảm ứng mạnh của heme oxyase-1, được chứng minh là có tác dụng bảo vệ tế bào chống lại tổn thương thận [68].
Con đường MAPK cũng liên quan đến tổn thương thận do oxy hóa. Kích hoạt tín hiệu ngoại bào điều tiết kinase (ERK) hoặc ức chế JNK đã được chứng minh là làm giảm tổn thương thận do oxy hóa cái mà gây ra hoại tử các tế bào ống thận gần của chuột trong thực nghiệm [69]. Sự điều hòa của ERK cũng có vai trò quan trọng trong hiệu quả sớm, theo đó, ngăn chặn thiếu máu cục bộ sớm có tác dụng bảo vệ chống lại tổn thương thiếu máu cục bộ/tái tưới máu sau đó [70].
Thay đổi trong chu kỳ tế bào cũng đã được chứng minh là có liên quan đến tổn thương thiếu máu cục bộ thận. Tăng điều hòa của p21, dường như cho phép sửa chữa và tái tạo tế bào, trong khi chuột được loại bỏ p21 cho thấy hoại tử tế bào tăng khi đáp ứng với thiếu máu cục bộ [71].
Điều trị
Điều trị thay thế thận trong suy thận cấp thường bao gồm thận nhân tạo cách quãng (IHD) hoặc điều trị thay thế thận liên tục (CRRT), ví dụ, lọc máu tĩnh - tĩnh mạch liên tục (CVVH). Do CVVH ít gây biến đổi huyết động hơn so với IHD nên sử dụng CVVH có lợi hơn là IHD, vì bất kỳ sự thay đổi huyết động hoặc nhiễm độc thận nào bổ sung, có thể làm kéo dài quá trình suy thận cấp và do đó làm tăng tỷ lệ tử vong. Tuy nhiên, những nghiên cứu gần đây nhất về kết quả so sánh ngẫu nhiên IHD với CRRT trong suy thận cấp không cho thấy bất kỳ sự khác biệt nào về tỷ lệ sống sót giữa hai phương pháp [72].
Cũng như với hầu hết các tình trạng bệnh lý khác, bệnh nhân thiếu máu cục bộ thận cấp được can thiệp càng sớm kết quả càng thuận lợi. Các dấu ấn sinh học nhạy hơn giúp cho chẩn đoán sớm so với sự gia tăng nồng độ creatinine huyết thanh ở bệnh nhân suy thận cấp cần được nghiên cứu để có thể giúp chẩn đoán sớm và can thiệp sớm. Như đã thảo luận ở trên, có một số dấu hiệu chẩn đoán đang được nghiên cứu. Tuy nhiên, hiện nay, việc xác định FE Na bằng cách sử dụng các phép đo natri và creatinine trong nước tiểu tại chỗ là dấu hiệu ban đầu và sẵn có nhất để chẩn đoán sớm suy thận cấp.
Những bệnh nhân có thời gian lâm sàng suy thận cấp kéo dài và cần phải lọc máu là những yếu tố chính dự báo tiên lượng xấu. Bệnh nhân mắc suy thận cấp cần phải lọc máu có tỷ lệ tử vong 50% - 70%. Nhiễm khuẩn và biến chứng tim phổi là nguyên nhân chính gây tử vong ở bệnh nhân bị suy thận cấp. Truyền dịch quá mức ở những bệnh nhân đã bị suy thận cấp có thể dẫn đến xung huyết phổi, thiếu oxy, cần hỗ trợ thở máy, viêm phổi và hội chứng suy đa tạng có tỷ lệ tử vong 80% - 90% [9, 73, 74]. Cho đến khi các phương pháp điều trị có thể làm đảo ngược được các rối loạn ở bệnh nhân suy thận cấp để bảo vệ bệnh nhân, thì mọi nỗ lực nên được thực hiện như đặt ống thông bàng quang, đường truyền tĩnh mạch và thở máy, để tránh phải sử dụng các thủ thuật xâm lấn.
Cùng với sự chăm sóc hỗ trợ như trên, cần kết hợp điều trị các yếu tố làm giảm mức lọc cầu thận để ngăn ngừa hoặc làm giảm quá trình tiến triển của suy thận cấp. Liệu pháp kết hợp như vậy liên quan đến các thuốc có lợi cho trương lực mạch máu, tắc nghẽn ống thận và viêm. Tuy nhiên, các thuốc giãn mạch như thuốc chẹn kênh calci và thuốc lợi tiểu thải natri có thể gây ra các tác dụng không mong muốn như giãn mạch toàn thân và hạ huyết áp [75, 76], làm tăng trương lực giao cảm và tăng hoạt động của hệ thống renin-angiotensin [77]. Hormon thần kinh này có tác dụng hỗ trợ huyết áp nhưng gây co mạch thận [78], có thể làm mờ tác dụng có lợi của thuốc chẹn kênh calci và peptide lợi tiểu thải natri ở thận.
Chỉ cần cải thiện mức lọc cầu thận 10 ml/phút từ mỗi thận ở bệnh nhân bị suy thận cấp có thể tránh được nhu cầu lọc máu và có thể cải thiện khả năng sống sót, nên đưa vào hai bên thận thuốc giãn mạch tác dụng ngắn và/hoặc chất lợi tiểu thẩm thấu như mannitol là cách tiếp cận ít xâm lấn hơn là sử dụng thận nhân tạo [79]. Một cách tiếp cận khác là kết hợp một thuốc giãn mạch toàn thân như peptide lợi tiểu thải natri, với dopamine [80] hoặc mannitol [81], điều này không chỉ làm giảm tác dụng hạ huyết áp toàn thân của thuốc giãn mạch mà còn có thể làm tăng lưu lượng dịch trong lòng ống thận. Phương pháp điều trị kết hợp này đã được chứng minh là có hiệu quả trong tổn thương thận cấp tính ở động vật thực nghiệm [81].
Tác dụng của các chất chống viêm, bao gồm cả các chất có tác dụng loại trừ gốc oxy, trong điều trị suy thận cấp cũng cần được nghiên cứu. Như đã thảo luận trên đây, việc sử dụng các chất ức chế NOS phải cụ thể, vì suy thận cấp trở nên nghiêm trọng hơn do ức chế NOS không đặc hiệu [66]. Ví dụ, một chất ức chế iNOS cụ thể, L -NIL, đã được chứng minh là đủ khả năng bảo vệ thận trong suy thận cấp liên quan đến nhiễm trùng huyết ở chuột [67]. Về vấn đề này, tổn thương ống thận do cytokine gây ra có thể xảy ra trong nhiễm trùng huyết trong trường hợp không giảm lưu lượng máu thận. Tác dụng chống viêm và ức chế iNOS của αMSH, được chứng minh là có hiệu lực đến 8 giờ sau khi sử dụng. Vấn đề này cần được nghiên cứu như một cách tiếp cận để thay đổi quá trình điều trị suy thận cấp lâm sàng [61]. Sự ức chế TNF-a đủ khả năng bảo vệ thận trong suy thận cấp do nội độc tố trong thực nghiệm [82]. Tuy nhiên, việc sử dụng kháng thể kháng TNF-α ở bệnh nhân nhiễm trùng không thấy cải thiện tỷ lệ sống sót [83]. Trong thiếu máu cục bộ thận cấp, có sự phân hủy ATP xảy ra cùng với sự rò rỉ các sản phẩm nucleotide ra khỏi tế bào ống thận. Trên nền tảng này, việc cung cấp ATP ngoại sinh đã được chứng minh là có khả năng bảo vệ thận trong thiếu máu cục bộ thận trên thực nghiệm. Trong khi ATP đã được chứng minh là bảo vệ chống lại hoại tử ống thận trong thực nghiệm [84], bằng chứng thực nghiệm gần đây cho thấy GTP có thể có tác dụng ngăn ngừa chết tế bào theo chương trình liên quan đến tổn thương thận cấp tính [85]. Hiệu quả của việc sử dụng peptide RGD tổng hợp để làm giảm sự tắc nghẽn ống thận vẫn chưa được kiểm chứng trong suy thận cấp lâm sàng. Tuy nhiên, vì bệnh nhân thường được quan sát thấy sau khi bị suy thận cấp lâm sàng với mức lọc cầu thận giảm dưới 10% so với bình thường, việc cung cấp các peptide RGD tổng hợp này vào lòng ống thận sẽ khó khăn trong trường hợp không đi kèm giãn mạch thận. Các nghiên cứu thực nghiệm kiểm tra sự điều hòa của protein sốc nhiệt [85, 86] hoặc tác dụng bảo vệ chống lại tổn thương sau này [70] cho thấy các phương pháp bảo vệ thận cần được nghiên cứu thêm.
Vì bệnh nhân bị suy thận cấp lâm sàng thường được quan sát thấy sau khi có nguyên nhân tác động, ngoại trừ đối tượng ghép thận [22] và người được điều trị bằng phóng xạ [87], các phương pháp giúp phục hồi và làm giảm thời gian điều trị suy thận cấp lâm sàng đã được đề cập ở trên. Thật không may, tác dụng có lợi của yếu tố tăng trưởng có nguồn gốc từ insulin được quan sát thấy ở động vật thực nghiệm lại không thấy trong một nghiên cứu ngẫu nhiên ở bệnh nhân mắc suy thận cấp lâm sàng [88]. Một cách tiếp cận khác gần đây trong lĩnh vực này là sử dụng tế bào gốc hoặc tế bào nội mô để tăng cường phục hồi sau tổn thương thận do thiếu máu cục bộ cấp tính ở động vật thực nghiệm [89 – 92]. Hiện đang có nghiên cứu về các cơ chế trong đó các ống thận bị tổn thương được liên kết với các tế bào mới tích cực tham gia tổng hợp DNA. Các con đường mà các tế bào sống sót tái lập chu kỳ tế bào và sao chép có thể liên quan đến các gen phản ứng ngay lập tức sớm [93].
Tóm lại, trong khi chúng ta đã biết về tổn thương thiếu máu cục bộ thận cấp, thường gặp ở bệnh nhân nhập viện, tỷ lệ tử vong của suy thận cấp lâm sàng vẫn còn cao. Tuy nhiên, tương lai có nhiều hứa hẹn cho chẩn đoán sớm và các can thiệp hiệu quả có khả năng ngăn ngừa hoặc rút ngắn quá trình tổn thương thận cấp tính và do đó cải thiện khả năng sống sót. Một mạng lưới các thử nghiệm lâm sàng với suy thận cấp cùng với những thống kê đầy đủ chắc chắn sẽ tạo điều kiện thuận lợi cho quá trình này.
Tài liệu tham khảo:
1. Bywaters EG, Beall D. Crush injuries with impairment of renal function. Br. Med. J. 1941;1:427–432.[PMC free article] [PubMed]
2. Teschan PE, et al. Post-traumatic renal insufficiency in military casualties. I. Clinical characteristics. Am. J. Med. 1955;18:172–186. [PubMed]
3. Smith LH, Jr, et al. Post-traumatic renal insufficiency in military casualties. II. Management, use of an artificial kidney, prognosis. Am. J. Med. 1955;18:187–198. [PubMed]
4. Butkus DE. Post-traumatic acute renal failure in combat casualties: a historical review. Mil. Med.1984;149:117–124. [PubMed]
5. Whelton A, Donadio JV., Jr Post-traumatic acute renal failure in Vietnam. A comparison with the Korean war experience. Johns Hopkins Med. J. 1969;124:95–105. [PubMed]
6. Reineck HJ, O’Connor GJ, Lifschitz MD, Stein JH. Sequential studies on the pathophysiology of glycerol-induced acute renal failure. J. Lab. Clin. Med. 1980;96:356–362. [PubMed]
7. Miller TR, et al. Urinary diagnostic indices in acute renal failure: a prospective study. Ann. Intern. Med.1978;89:47–50. [PubMed]
8. Carvounis CP, Nisar S, Guro-Razuman S. Significance of the fractional excretion of urea in the differential diagnosis of acute renal failure. Kidney Int. 2002;62:2223–2229. [PubMed]
9. Esson ML, Schrier RW. Diagnosis and treatment of acute tubular necrosis. Ann. Intern. Med.2002;137:744–752. [PubMed]
10. Mehta RL, et al. Nephrology consultation in acute renal failure: does timing matter? Am. J. Med.2002;113:456–461. [PubMed]
11. Coll E, et al. Serum cystatin C as a new marker for noninvasive estimation of glomerular filtration rate and as a marker for early renal impairment. Am. J. Kidney Dis. 2000;36:29–34. [PubMed]
12. Han WK, Bailly V, Abichandani R, Thadhani R, Bonventre JV. Kidney Injury Molecule-1 (KIM-1): a novel biomarker for human renal proximal tubule injury. Kidney Int. 2002;62:237–244. [PubMed]
13. Parikh CR, Jani A, Melnikov VY, Faubel S, Edelstein CL. Urinary interleukin-18 is a marker of human acute tubular necrosis. Am. J. Kidney Dis. 2004;43:405–414. [PubMed]
14. Westhuyzen J, et al. Measurement of tubular enzymuria facilitates early detection of acute renal impairment in the intensive care unit. Nephrol. Dial. Transplant. 2003;18:543–551. [PubMed]
15. Kribben A, Edelstein CL, Schrier RW. Pathophysiology of acute renal failure. J. Nephrol.1999;12(Suppl. 2):S142–S151. [PubMed]
16. Racusen, L.C., and Nast, C.C. 1999. Renal histopathology, urine cytology, and cytopathology in acute renal failure. In Atlas of diseases of the kidney. R.W. Schrier, editor. Blackwell Science. Philadelphia, Pennsylvania, USA. 1–9, 12.
17. Conger JD, Robinette JB, Schrier RW. Smooth muscle calcium and endothelium-derived relaxing factor in the abnormal vascular responses of acute renal failure. J. Clin. Invest. 1988;82:532–537.[PMC free article] [PubMed]
18. Conger JD, Falk SA. Abnormal vasoreactivity of isolated arterioles from rats with ischemic acute renal (ARF) [abstract] J. Am. Soc. Nephrol. 1993;4:733A.
19. Burke TJ, et al. Protective effect of intrarenal calcium membrane blockers before or after renal ischemia. Functional, morphological, and mitochondrial studies. J. Clin. Invest. 1984;74:1830–1841.[PMC free article] [PubMed]
20. Arnold PE, Lumlertgul D, Burke TJ, Schrier RW. In vitro versus in vivo mitochondrial calcium loading in ischemic acute renal failure. Am. J. Physiol. 1985;248:F845–F850. [PubMed]
21. Arnold PE, Van Putten VJ, Lumlertgul D, Burke TJ, Schrier RW. Adenine nucleotide metabolism and mitochondrial Ca2+ transport following renal ischemia. Am. J. Physiol. 1986;250:F357–F363. [PubMed]
22. Neumayer HH, Wagner K. Prevention of delayed graft function in cadaver kidney transplants by diltiazem: outcome of two prospective, randomized clinical trials. J. Cardiovasc. Pharmacol.1987;10(Suppl. 10):S170–S177. [PubMed]
23. Mason J, Torhorst J, Welsch J. Role of the medullary perfusion defect in the pathogenesis of ischemic renal failure. Kidney Int. 1984;26:283–293. [PubMed]
24. Kelly KJ, Williams WW, Jr, Colvin RB, Bonventre JV. Antibody to intercellular adhesion molecule 1 protects the kidney against ischemic injury. Proc. Natl. Acad. Sci. U. S. A. 1994;91:812–816.[PMC free article] [PubMed]
25. Molitoris BA, Marrs J. The role of cell adhesion molecules in ischemic acute renal failure. Am. J. Med.1999;106:583–592. [PubMed]
26. Zizzi HC, et al. Quantification of P-selectin expression after renal ischemia and reperfusion. J. Pediatr. Surg. 1997;32:1010–1013. [PubMed]
27. Singbartl K, Green SA, Ley K. Blocking P-selectin protects from ischemia/reperfusion-induced acute renal failure. FASEB J. 2000;14:48–54. [PubMed]
28. Linas SL, Shanley PF, Whittenburg D, Berger E, Repine JE. Neutrophils accentuate ischemia-reperfusion injury in isolated perfused rat kidneys. Am. J. Physiol. 1988;255:F728–F735. [PubMed]
29. Molitoris BA, Sandoval R, Sutton TA. Endothelial injury and dysfunction in ischemic acute renal failure. Crit. Care Med. 2002;30(Suppl. 5):S235–S240. [PubMed]
30. Sutton TA, Fisher CJ, Molitoris BA. Microvascular endothelial injury and dysfunction during ischemic acute renal failure. Kidney Int. 2002;62:1539–1549. [PubMed]
31. Badr KF, et al. Mesangial cell, glomerular and renal vascular responses to endothelin in the rat kidney. Elucidation of signal transduction pathways. J. Clin. Invest. 1989;83:336–342. [PMC free article][PubMed]
32. Thadhani R, Pascual M, Bonventre JV. Acute renal failure. N. Engl. J. Med. 1996;334:1448–1460.[PubMed]
33. Racusen LC. Epithelial cell shedding in acute renal injury. Clin. Exp. Pharmacol. Physiol.1998;25:273–275. [PubMed]
34. Molitoris BA, Chan LK, Shapiro JI, Conger JD, Falk SA. Loss of epithelial polarity: a novel hypothesis for reduced proximal tubule Na+ transport following ischemic injury. J. Membr. Biol. 1989;107:119–127.[PubMed]
35. Alejandro VS, et al. Postischemic injury, delayed function and Na+/K(+)-ATPase distribution in the transplanted kidney. Kidney Int. 1995;48:1308–1315. [PubMed]
36. Edelstein, C., and Schrier, R. 1999. The role of calpain in renal proximal tubular and hepatocyte injury. In Calpain: pharmacology and toxicology of calcium-dependent protease. K.K. Wang and P.W. Yuen, editors. Taylor & Francis. Philadelphia, Pennsylvania, USA. 307–329.
37. Kribben A, et al. Evidence for role of cytosolic free calcium in hypoxia-induced proximal tubule injury. J. Clin. Invest. 1994;93:1922–1929. [PMC free article] [PubMed]
38. Edelstein CL, et al. The role of cysteine proteases in hypoxia-induced rat renal proximal tubular injury. Proc. Natl. Acad. Sci. U. S. A. 1995;92:7662–7666. [PMC free article] [PubMed]
39. Edelstein CL, et al. Effect of glycine on prelethal and postlethal increases in calpain activity in rat renal proximal tubules. Kidney Int. 1997;52:1271–1278. [PubMed]
40. Shi Y, Melnikov VY, Schrier RW, Edelstein CL. Downregulation of the calpain inhibitor protein calpastatin by caspases during renal ischemia-reperfusion. Am. J. Physiol. Renal Physiol. 2000;279:F509–F517. [PubMed]
41. Choi KH, Edelstein CL, Gengaro P, Schrier RW, Nemenoff RA. Hypoxia induces changes in phospholipase A2 in rat proximal tubules: evidence for multiple forms. Am. J. Physiol. 1995;269:F846–F853. [PubMed]
42. Oliver J, Mac DM, Tracy A. The pathogenesis of acute renal failure associated with traumatic and toxic injury: renal ischemia, nephrotoxic damage and the ischemic episode. J. Clin. Invest. 1951;30:1307–1439.[PMC free article] [PubMed]
43. Wangsiripaisan A, Gengaro PE, Edelstein CL, Schrier RW. Role of polymeric Tamm-Horsfall protein in cast formation: oligosaccharide and tubular fluid ions. Kidney Int. 2001;59:932–940. [PubMed]
44. Zuk A, Bonventre JV, Matlin KS. Expression of fibronectin splice variants in the postischemic rat kidney. Am. J. Physiol. Renal Physiol. 2001;280:F1037–F1053. [PubMed]
45. Tanner GA. Nephron obstruction and tubuloglomerular feedback. Kidney Int. Suppl. 1982;12:S213–S218. [PubMed]
46. Conger JD, Robinette JB, Kelleher SP. Nephron heterogeneity in ischemic acute renal failure. Kidney Int. 1984;26:422–429. [PubMed]
47. Noiri E, et al. Cyclic RGD peptides ameliorate ischemic acute renal failure in rats. Kidney Int.1994;46:1050–1058. [PubMed]
48. Goligorsky MS, DiBona GF. Pathogenetic role of Arg-Gly-Asp-recognizing integrins in acute renal failure. Proc. Natl. Acad. Sci. U. S. A. 1993;90:5700–5704. [PMC free article] [PubMed]
49. Burke TJ, Cronin RE, Duchin KL, Peterson LN, Schrier RW. Ischemia and tubule obstruction during acute renal failure in dogs: mannitol in protection. Am. J. Physiol. 1980;238:F305–F314. [PubMed]
50. Schnermann J. Homer W. Smith Award lecture. The juxtaglomerular apparatus: from anatomical peculiarity to physiological relevance. J. Am. Soc. Nephrol. 2003;14:1681–1694. [PubMed]
51. Molitoris BA, Falk SA, Dahl RH. Ischemia-induced loss of epithelial polarity. Role of the tight junction. J. Clin. Invest. 1989;84:1334–1339. [PMC free article] [PubMed]
52. Edelstein, C., and Schrier, R. 2001. Pathophysiology of ischemic acute renal failure. In Diseases of the kidney and urinary tract. 7th edition. R.W. Schrier, editor. Lippincott Williams & Wilkins. Philadelphia, Pennsylvania, USA. 1041–1070.
53. Myers BD, Chui F, Hilberman M, Michaels AS. Transtubular leakage of glomerular filtrate in human acute renal failure. Am. J. Physiol. 1979;237:F319–F325. [PubMed]
54. Yu L, Gengaro PE, Niederberger M, Burke TJ, Schrier RW. Nitric oxide: a mediator in rat tubular hypoxia/reoxygenation injury. Proc. Natl. Acad. Sci. U. S. A. 1994;91:1691–1695. [PMC free article][PubMed]
55. Noiri E, Peresleni T, Miller F, Goligorsky MS. In vivo targeting of inducible NO synthase with oligodeoxynucleotides protects rat kidney against ischemia. J. Clin. Invest. 1996;97:2377–2383.[PMC free article] [PubMed]
56. Ling H, et al. Effect of hypoxia on proximal tubules isolated from nitric oxide synthase knockout mice. Kidney Int. 1998;53:1642–1646. [PubMed]
57. Ling H, et al. Attenuation of renal ischemia-reperfusion injury in inducible nitric oxide synthase knockout mice. Am. J. Physiol. 1999;277:F383–F390. [PubMed]
58. Xia Y, Dawson VL, Dawson TM, Snyder SH, Zweier JL. Nitric oxide synthase generates superoxide and nitric oxide in arginine-depleted cells leading to peroxynitrite-mediated cellular injury. Proc. Natl. Acad. Sci. U. S. A. 1996;93:6770–6774. [PMC free article] [PubMed]
59. Noiri E, et al. Oxidative and nitrosative stress in acute renal ischemia. Am. J. Physiol. Renal Physiol.2001;281:F948–F957. [PubMed]
60. Wangsiripaisan A, et al. Effect of nitric oxide donors on renal tubular epithelial cell-matrix adhesion. Kidney Int. 1999;55:2281–2288. [PubMed]
61. Chiao H, et al. Alpha-melanocyte-stimulating hormone protects against renal injury after ischemia in mice and rats. J. Clin. Invest. 1997;99:1165–1172. [PMC free article] [PubMed]
62. Wang W, et al. Interaction among nitric oxide, reactive oxygen species, and antioxidants during endotoxemia-related acute renal failure. Am. J. Physiol. Renal Physiol. 2003;284:F532–F537. [PubMed]
63. Melnikov VY, et al. Impaired IL-18 processing protects caspase-1-deficient mice from ischemic acute renal failure. J. Clin. Invest. 2001;107:1145–1152. [PMC free article] [PubMed]
64. Melnikov VY, et al. Neutrophil-independent mechanisms of caspase-1– and IL-18–mediated ischemic acute tubular necrosis in mice. J. Clin. Invest. 2002;110:1083–1091. doi:10.1172/JCI200215623.[PMC free article] [PubMed]
65. Wang, W., et al. 2004. Endothelial nitric oxide synthase (eNOS) deficient mice exhibit increased susceptibility to endotoxin-induced acute renal failure. Am. J. Physiol. In press. [PubMed]
66. Atanasova I, Burke TJ, McMurtry IF, Schrier RW. Nitric oxide synthase inhibition and acute renal ischemia: effect on systemic hemodynamics and mortality. Ren. Fail. 1995;17:389–403. [PubMed]
67. Schwartz D, et al. Inhibition of constitutive nitric oxide synthase (NOS) by nitric oxide generated by inducible NOS after lipopolysaccharide administration provokes renal dysfunction in rats. J. Clin. Invest.1997;100:439–448. [PMC free article] [PubMed]
68. Sikorski EM, Hock T, Hill-Kapturczak N, Agarwal A. The story so far: molecular regulation of the heme oxygenase-1 gene in renal injury. Am. J. Physiol. Renal Physiol. 2004;286:F425–F441. [PubMed]
69. Arany I, Megyesi JK, Kaneto H, Tanaka S, Safirstein RL. Activation of ERK or inhibition of JNK ameliorates H2O2 cytotoxicity in mouse renal proximal tubule cells. Kidney Int. 2004;65:1231–1239.[PubMed]
70. Park KM, Chen A, Bonventre JV. Prevention of kidney ischemia/reperfusion-induced functional injury and JNK, p38, and MAPK kinase activation by remote ischemic pretreatment. J. Biol. Chem.2001;276:11870–11876. [PubMed]
71. Price PM, Megyesi J, Safirstein RL. Cell cycle regulation: repair and regeneration in acute renal failure. Semin. Nephrol. 2003;23:449–459. [PubMed]
72. Teehan GS, et al. Dialysis membrane and modality in acute renal failure: understanding discordant meta-analyses. Semin. Dial. 2003;16:356–360. [PubMed]
73. Brivet FG, Kleinknecht DJ, Loirat P, Landais PJ. Acute renal failure in intensive care units: causes, outcome, and prognostic factors of hospital mortality; a prospective, multicenter study. French Study Group on Acute Renal Failure. Crit. Care Med. 1996;24:192–198. [PubMed]
74. Poole, B., and Schrier, R. 2005. Acute renal failure in the intensive care unit. In Textbook of critical care. 5th edition. F. Mitchell, E. Abraham, J.Vincent, and P. Kochanek, editors. Elsevier. Philadelphia, Pennsylvania, USA. In press.
75. Nakamoto M, Shapiro JI, Shanley PF, Chan L, Schrier RW. In vitro and in vivo protective effect of atriopeptin III on ischemic acute renal failure. J. Clin. Invest. 1987;80:698–705. [PMC free article][PubMed]
76. Allgren RL, et al. Anaritide in acute tubular necrosis. Auriculin Anaritide Acute Renal Failure Study Group. N. Engl. J. Med. 1997;336:828–834. [PubMed]
77. Schrier R, Wang W. Acute renal failure and sepsis. N. Engl. J. Med. 2004;351:35–45.
78. Wang W, et al. Protective effect of renal denervation on normotensive endotoxemia-induced acute renal failure in mice. Am. J. Physiol. Renal Physiol. 2002;283:F583–F587. [PubMed]
79. Burke TJ, Arnold PE, Schrier RW. Prevention of ischemic acute renal failure with impermeant solutes. Am. J. Physiol. 1983;244:F646–F649. [PubMed]
80. Conger JD, Falk SA, Yuan BH, Schrier RW. Atrial natriuretic peptide and dopamine in a rat model of ischemic acute renal failure. Kidney Int. 1989;35:1126–1132. [PubMed]
81. Lieberthal W, Sheridan AM, Valeri CR. Protective effect of atrial natriuretic factor and mannitol following renal ischemia. Am. J. Physiol. 1990;258:F1266–F1272. [PubMed]
82. Knotek M, et al. Endotoxemic renal failure in mice: role of tumor necrosis factor independent of inducible nitric oxide synthase. Kidney Int. 2001;59:2243–2249. [PubMed]
83. Abraham E, et al. Double-blind randomised controlled trial of monoclonal antibody to human tumour necrosis factor in treatment of septic shock. NORASEPT II Study Group. Lancet. 1998;351:929–933.[PubMed]
84. Siegel NJ, et al. Enhanced recovery from acute renal failure by the postischemic infusion of adenine nucleotides and magnesium chloride in rats. Kidney Int. 1980;17:338–349. [PubMed]
85. Kelly KJ, Plotkin Z, Dagher PC. Guanosine supplementation reduces apoptosis and protects renal function in the setting of ischemic injury. J. Clin. Invest. 2001;108:1291–1298. doi:10.1172/JCI200113018.[PMC free article] [PubMed]
86. Meldrum KK, Meldrum DR, Sezen SF, Crone JK, Burnett AL. Heat shock prevents simulated ischemia-induced apoptosis in renal tubular cells via a PKC-dependent mechanism. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2001;281:R359–R364. [PubMed]
87. Tepel M, et al. Prevention of radiographic-contrast-agent-induced reductions in renal function by acetylcysteine. N. Engl. J. Med. 2000;343:180–184. [PubMed]
88. Hirschberg R, et al. Multicenter clinical trial of recombinant human insulin-like growth factor I in patients with acute renal failure. Kidney Int. 1999;55:2423–2432. [PubMed]
89. Brodsky SV, et al. Endothelial dysfunction in ischemic acute renal failure: rescue by transplanted endothelial cells. Am. J. Physiol. Renal Physiol. 2002;282:F1140–F1149. [PubMed]
90. Kale S, et al. Bone marrow stem cells contribute to repair of the ischemically injured renal tubule. J. Clin. Invest. 2003;112:42–49. doi:10.1172/JCI200317856. [PMC free article] [PubMed]
91. Lin F, et al. Hematopoietic stem cells contribute to the regeneration of renal tubules after renal ischemia-reperfusion injury in mice. J. Am. Soc. Nephrol. 2003;14:1188–1199. [PubMed]
92. Gupta S, Verfaillie C, Chmielewski D, Kim Y, Rosenberg ME. A role for extrarenal cells in the regeneration following acute renal failure. Kidney Int. 2002;62:1285–1290. [PubMed]
93. Safirstein R, DiMari J, Megyesi J, Price P. Mechanisms of renal repair and survival following acute injury. Semin. Nephrol. 1998;18:519–522. [PubMed]
94. Thadhani R, Pascual M, Bonventre J. Acute renal failure. N. Engl. J. Med. 1996;334:1448–1460.[PubMed]



















