





Bệnh tan máu bẩm sinh (Bệnh Thalassemia)
PGS.TS. Hà Hoàng Kiệm, bệnh viện 103, Học viện Quân y.
1. Đại cương
1.1. Khái niệm
Bệnh bệnh tan máu bẩm sinh còn gọi là bệnh Thalassemia, là một bệnh di truyền về máu có liên quan đến đột biến gen quy định sản sinh ra huyết sắc tố (hemoglobin). Đột biến gen làm tạo ra huyết sắc tố bất thường dẫn đến hồng cầu dễ bị vỡ (bị tan) sớm. Kết quả là bệnh nhân không đủ hồng cầu để vận chuyển oxy gây ra thiếu máu. Việc hồng cầu vỡ nhiều làm giải phóng ra quá nhiều sắt vào máu nên gây ra tình trạng tích lũy sắt ở các nội tạng, tình trạng nhiễm sắt này nếu kéo dài có thể gây độc cho cơ thể.
1.2. Dịch tễ
Mỗi năm, trên thế giới có khoảng 100.000 trẻ em sinh ra mắc bệnh Thalassemia. Bệnh gặp nhiều ở Ý, Hy Lạp, Trung Đông, Nam Á và châu Phi.
Bệnh tan Thalassiemia hiện diện tại mọi quốc gia và dân tộc, xuất hiện ở cả nam và nữ. Hiện nay, ước tính khoảng 7% dân số thế giới mang gen bệnh, 1,1% các cặp vợ chồng có nguy cơ sinh con bị bệnh, 0,27% trường hợp có thai sinh ra con bị bệnh, mỗi năm có khoảng 300.000 - 500.000 trẻ sinh ra bị bệnh Thalassemia mức độ nặng. Tại Việt Nam hiện nay, ước tính có khoảng 20.000 người bị bệnh tan máu bẩm sinh thể nặng, hàng năm có thêm khoảng 2.000 trẻ em sinh ra bị bệnh tan máu bẩm sinh và khoảng 10 - 12 triệu người đang mang gen bệnh Thalassemia (người mang gen thể lặn không có biểu hiện bệnh lý nhưng là nguồn di truyền gen bệnh cho thế hệ sau). Do đó, vấn đề tầm soát Thalassemia tiền hôn nhân trở nên quan trọng trong việc phòng ngừa cũng như chuẩn bị cho việc mang thai. Thalassemia là một căn bệnh di truyền nguy hiểm, khó chữa, nhưng lại dễ phòng ngừa thông qua xét nghiệm sàng lọc, phát hiện gen bệnh ở giai đoạn tiền hôn nhân hoặc chẩn đoán trước sinh.
2. Nguyên nhân gây bệnh
Bệnh Thalassemia là do cấu tạo không bình thường của hemoglobin trong hồng cầu. Hb người bình thường là HbA (Adult), chiếm hơn 97% trong máu, gồm 2 chuỗi globin α và 2 chuỗi globin β (α2β2). Chuỗi α có 141 acid amin và chuỗi β có 146 acid amin, trật tự sắp xếp các acid amin trong các chuỗi này là tuyệt đối, chỉ cần thay đổi một vị trí là Hb sẽ bị biến tính làm hình dạng hồng cầu dễ thay đổi và dễ bị vỡ gây ra các bệnh lý thiếu máu và tan máu. Do vậy, bệnh Thalassemia có hai dạng bất thường chính được gọi là alpha-Thalassemia và beta -Thalassemia, tùy theo phần nào của chất hemoglobin bị thiếu. (Hemoglobin - Hb, còn gọi là huyết sắc tố, là một protein màu - chromoprotein, gồm hai thành phần là nhân heme và globin. Heme là một sắc tố đỏ. Mỗi heme gồm một vòng porphyrin và một Fe2+ chính giữa. Một phân tử hemoglobin gồm có 4 nhân heme, chiếm 5% trọng lượng của phân tử Hb. Globin là một protein gồm 4 chuỗi polypeptid giống nhau từng đôi một, đó là 2 chuỗi alpha và 2 chuỗi beta).
- Dạng alpha-thalassemia:
Dạng bệnh này có thể gặp thiếu hụt 1, hoặc cả 2 chuỗi alpha-globin. Thiếu hụt 2 chuỗi alpha-globin là nặng. Thể nặng của alpha-thalassemia, gặp nhiều nhất trong các dân tộc ở Đông Nam Á, Trung Quốc và Philippines, thường gây hư thai hay trẻ em chết khi sinh. Phần lớn những trẻ em có bệnh alpha-thalassemia bị thiếu máu mạn tính, một số bị nặng trầm trọng, không sống được đến tuổi trưởng thành.
- Dạng beta-thalassemia:
Loại bệnh này cũng bao gồm thiếu hụt 1 hoặc thiếu hụt 2 chuỗi beta-globin. Do đó lâm sàng cũng gặp một số trường hợp từ nặng đến nhẹ, một số trường hợp không thấy ảnh hường đến sức khỏe.
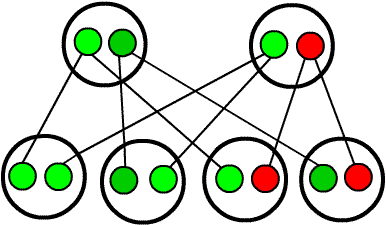
Gen Thalassemia tồn tại ở dạng cặp, gồm 2 gen (A hoặc a). Người khỏe mạnh có 2 gen trội (AA). Người bị bệnh có 2 gen lặn (aa). Người có 1 gen trội và 1 gen lặn thành thể di hợp (Aa) sẽ không có biểu hiện bệnh, nhưng khi kết hôn với người mang thể dị hợp khác (Aa) thì con sinh ra có 25% bị bệnh (aa).
.jpg)
Hình 1. Kiểu di truyền bố hoặc mẹ mang gen bệnh. Màu xanh: gen bình thường, màu đỏ: gen bệnh.
Bảng 1. Kiểu gen của người bình thường, người bệnh Thalassemia và người mang gen Thalassemia ẩn.
Người khỏe mạnhMang cả 2 gen Thalassemia ở dạng đồng hợp trội là AA, hoàn toàn khỏe mạnh và không có biểu hiện bệnh. Khi kết hôn với người khỏe mạnh (AA), bệnh (aa) hoặc mang gen dị hợp (Aa) thì sinh con ra khỏe mạnh (AA) hoặc bình thường nhưng mang gen dị hợp (Aa). |
Người bệnhMang cả 2 gen Thalassemia ở dạng đồng hợp lặn là aa, có biểu hiện bệnh thiếu máu rõ ràng. Khi kết hôn với người khỏe mạnh (AA) thì con bình thường nhưng mang gen dị hợp (Aa), kết hôn với người dị hợp (Aa) thì 75% con mang bệnh(aa), kết hôn với người bệnh (aa) thì 100% con mang bệnh (aa). |
Người mang gen ẩnMang 2 gen Thalassemia ở dạng dị hợp (Aa), không có biểu hiện bệnh. Khi kết hôn với người khỏe mạnh (AA) thì con khỏe mạnh (AA) hoặc dị hợp (Aa), kết hôn với người bệnh (aa) thì 75% con mang bệnh (aa), kết hôn với người mang gen dị hợp (Aa) thì 25% con mang bệnh (aa). |
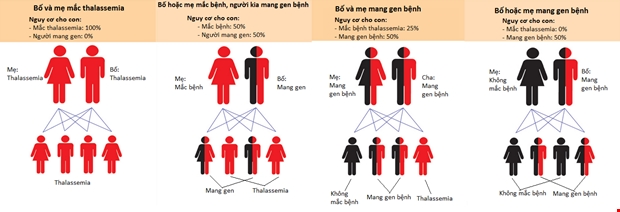
Hình 2. Các kiểu di truyền gen bệnh Thalassemia.
3. Lâm sàng
Do sự thiếu hụt tổng hợp một số chuỗi globin trong huyết sắc tố của hồng cầu, chất lượng hồng cầu suy giảm làm hồng cầu dễ bị vỡ (tan máu) dẫn đến thiếu máu mạn tính. Bệnh nhân mắc căn bệnh này hầu hết có biểu hiện giống nhau gồm 2 hội chứng: thiếu máu tan máu mạn tính và nhiễm sắt (nếu được truyền máu nhiều lần). Triệu chứng điển hình là thiếu máu, vàng mắt, sạm da, bụng trướng to, cùng với đó là hiện tượng phì đại xương gây biến dạng mặt, trán dô, mũi tẹt, răng hô, chậm phát triển… Biểu hiện chung của bệnh là:
- Mệt mỏi.
- Hoa mắt chóng mặt.
- Da xanh nhợt nhạt hơn bình thường.
- Da, củng mạc mắt vàng.
- Nước tiểu sẫm màu.
- Chậm lớn.
- Khó thở khi làm việc gắng sức….
3.1. Beta-thalassemia
Thiếu máu beta-thalassemia có hai loại nghiêm trọng, đó là thalassemia thể nặng và thalassemia thể trung gian (ít nghiêm trọng hơn). Thiếu máu trầm trọng có thể đe dọa đến tính mạng người bệnh. Các dấu hiệu và triệu chứng khác có thể bao gồm:
- Biếng ăn.
- Nhợt nhạt, da xanh xao.
- Dễ bị nhiễm khuẩn.
- Chán ăn.
- Không phát triển hoặc chậm phát triển.
- Vàng da: da có màu vàng, mắt không vàng.
- Các cơ quan bị to lên.
Loại bệnh thalassemia này thường nặng đến mức người bệnh đòi hỏi phải được truyền máu thường xuyên.
3.2. Alpha-thalassemia
Alpha-thalassemia cũng có hai loại nghiêm trọng, đó là rối loạn hemoglobin H và bệnh phù thai.
- Rối loạn hemoglobin H có thể gây ra các vấn đề về xương. Má, trán và hàm có thể phát triển quá mức. Ngoài ra, rối loạn hemoglobin H có thể gây ra:
+ Vàng da: da có màu vàng, mắt không vàng.
+ Lách to.
+ Dinh dưỡng kém.
- Bệnh phù thai được coi là một dạng thalassemia cực kỳ nghiêm trọng. Bệnh này xảy ra trước khi sinh. Hầu hết những trẻ bị bệnh này đều chết non hoặc chết ngay sau khi sinh.
3.3. Phân loại mức độ bệnh
Bệnh có 5 mức độ biểu hiện tùy theo số lượng gen bị tổn thương:
- Mức độ rất nặng: có biểu hiện phù thai từ khi còn trong bụng mẹ (những trường hợp này thường gây hỏng thai trước khi sinh).
- Mức độ nặng: có biểu hiện thiếu máu nặng khi trẻ chưa đến 2 tuổi.
- Mức độ trung bình: thường có biểu hiện thiếu máu rõ khi trẻ trên 6 tuổi.
- Mức độ nhẹ: triệu chứng máu thường rất kín đáo, người bệnh thường chỉ được phát hiện khi có kèm theo bệnh lý khác như nhiễm trùng, phẫu thuật, có thai…
- Thể ẩn: không có biểu hiện gì khác biệt, không thiếu máu (thậm chí có thể hiến máu được).
Nếu người bệnh không được điều trị sớm, đầy đủ sẽ xuất hiện nhiều biến chứng do thiếu máu và thừa sắt gây ra trên tất cả các cơ quan làm thay đổi diện mạo người bệnh như thể trạng thấp bé, trán dô, mũi tẹt, hàm răng hô, suy tim, suy gan, suy nội tiết..
Khi mới sinh ra, trẻ bị thể bệnh thalassemia nặng trông có vẻ khoẻ mạnh bình thường. Nhưng trong vòng vài tháng hay một hai năm, trẻ thường quấy khóc, mệt mỏi, biếng ăn, chậm lớn và da ửng màu vàng. Nếu không không được điều trị: lách, gan và tim sẽ to lên. Xương bị xốp và dễ gãy, cấu trúc của xương mặt bị thay đổi. Vì hồng cầu vỡ sớm hơn bình thường nên tủy xương phải làm việc quá sức (để sản xuất hồng cầu), khiến xương phì đại, biến dạng. Ở những trẻ bị Thalassemia nặng, trán gồ lên, mũi tẹt, xương hàm trên nhô ra do tăng sản tủy xương (trẻ em bị thalassemia nặng thường trông giống nhau vì cấu trúc xương mặt đều bị biến dạng tương tự). Trẻ sẽ chết sớm vì suy tim hay nhiễm khuẩn.
4. Điều trị
4.1. Một số phương pháp điều trị được sử dụng
- Truyền máu.
- Cấy ghép tủy xương (BMT).
- Thuốc và các chất bổ sung.
- Có thể phẫu thuật cắt bỏ lách hoặc túi mật.
4.2. Đối với bệnh nhân mức độ nặng và trung bình
- Truyền máu định kỳ và dùng thuốc thải sắt suốt cuộc đời.
- Đến khám và điều trị đúng hẹn.
- Khám lại ngay khi có dấu hiệu bất thường như: mệt nhiều, đau tim, khó thở, sốt cao, phù…
- Phẫu thuật cắt lách giúp kéo dài khoảng cách thời gian giữa các đợt truyền máu.
- Ghép tế bào gốc điều trị bệnh, biện pháp này cần phải đáp ứng điều kiện ngặt nghèo hơn như: phải tìm được nguồn tế bào gốc phù hợp, điều kiện sức khỏe đảm bảo, các chi phí ghép…
Truyền máu thường xuyên và sử dụng thuốc kháng sinh giúp trẻ bị thalassemia dạng trầm trọng có đời sống khả quan hơn. Truyền máu thường xuyên (một tuần 3-4 lần) có thể làm giảm bớt những biến chứng của bệnh (như suy tim hay xốp xương). Nhưng khi truyền máu quá nhiều như thế, sắt sẽ ứ đọng trong cơ thể, gây nhiễm sắt cho gan và tim.
Hiện nay, khi được điều trị, người bệnh thalassemia dạng trầm trọng có thể sống thêm từ 20 đến 30 năm.
Một số rất ít có thể chữa bằng phương pháp thay tủy.
4.3. Công tác điều dưỡng
Người bệnh Thalassemia có thể sinh hoạt, làm việc, kết hôn và sinh con khi được điều trị đầy đủ, tuân thủ các phác đồ điều trị và có chế độ dinh dưỡng, sinh hoạt hợp lý.
- Chế độ dinh dưỡng:
+ Cân bằng các chất giàu dinh dưỡng.
+ Không ăn thức ăn chứa nhiều sắt (thịt bò, mộc nhĩ, rau cải xoong…).
+ Để hạn chế hấp thu sắt khi ăn từ các thực phẩm sau bữa ăn nên uống 1 cốc nước chè xanh.
+ Bổ sung các thực phẩm giàu kẽm ( sò, củ cải đường, đậu nành...).
- Chế độ sinh hoạt:
+ Sinh hoạt bình thường , hạn chế lao động nặng các hoạt động gắng sức.
+ Tránh bị nhiễm trùng: rửa tay thường xuyên, tiêm vắc xin phòng bệnh.
+ Vận động, tập luyện các môn thể thao nhẹ nhàng, phù hợp…
5. Phòng bệnh
Để phòng bệnh cần hiểu biết về cơ chế di truyền. Tránh không sinh ra trẻ mang 2 gen bệnh do nhận từ cả bố và mẹ bằng các biện pháp như:
5.1. Tầm soát và phòng tránh bệnh từ sớm
Với các biện pháp xét nghiệm, tư vấn tiền hôn nhân. Các cặp vợ chồng chuẩn bị có thai hoặc đang mang thai, đặc biệt các gia đình đã có người bệnh Thalassemia nên được tư vấn và chẩn đoán tiền hôn nhân. Xét nghiệm phân tích gen Thalassemia gây bệnh thiếu máu của một cá thể, xác định rõ ràng tình trạng bệnh nặng hay nhẹ, hoặc không bị bệnh nhưng mang gen bệnh thể ẩn. Chỉ định xét nghiệm:
- Do tỉ lệ cao người mang gen bệnh (thể ẩn) trong quần thể, mọi cá nhân chuẩn bị kết hôn đều được khuyến khích thực hiện tầm soát bệnh Thalassemia trong xét nghiệm tiền hôn nhân để có kế hoạch thai sản tốt hơn. Nếu cả cha và mẹ đều có mang gen bệnh (thể ẩn) thì xác suất con bị bệnh Thalassemia lên đến 25%.
- Ngoài ra, các đối tượng sau cấp thiết cần làm tầm soát Thalassemia:
+ Vợ chồng đều có biểu hiện thiếu máu.
+ Vợ hoặc chồng mắc bệnh hoặc mang gen bệnh thể ẩn.
+ Công thức máu có hồng cầu nhỏ, dư sắt.
+ Trẻ sinh ra ốm yếu, xanh xao, vàng mắt, nhẹ cân, lách to.
+ Trẻ dễ mệt xuống sức khi vận động.
+ Gia đình có tiền sử bệnh thiếu máu.
5.2. Sàng lọc phát hiện bệnh sớm cho thai nhi
Phòng ngừa bằng các xét nghiệm tầm soát và chẩn đoán gen đột biến trong thai kỳ. Đây là biện pháp hiệu quả và chi phí thấp. Nếu cả vợ và chồng đều mang gen thì thai nhi có 25% nguy cơ bị mắc bệnh ở thể nặng, trường hợp này cần được thực hiện chẩn đoán trước sinh bằng phương pháp chọc ối hoặc sinh thiết gai nhau và tìm đột biến gen.
Đối với bệnh Thalassemia thai nhi, mẫu nước ối cần được thu để xét nghiệm. Trường hợp cha mẹ đều mang gen thể ẩn (gen bệnh) hoặc một trong hai người có bệnh Thalassemia, việc tầm soát bệnh Thalassemia cho thai là cần thiết và nên được thực hiện càng sớm càng tốt trong thai kỳ.
Thalassemia là bệnh có thể phòng tránh được. Do vậy, việc phòng tránh, tìm hiểu và được tư vấn, tầm soát gen bệnh sớm, trước kết hôn sẽ hạn chế được nguy cơ sinh ra những đứa trẻ mang gen hoặc bị bệnh, góp phần đảm bảo chất lượng dân số cho cộng đồng.
5.3. Những nghiên cứu đang tiến hành
- Tìm cách làm giảm độ ứ thừa của sắt trong cơ thể của bệnh nhân sau khi được truyền nhiều máu. Các nhà khoa học đang tìm và thử các chất có khả năng thu nhận sắt để giúp bài tiềt chất này ra khỏi cơ thể.
- Điều trị bằng gen, có thể có những phương hướng sau:
+ Tìm cách đưa một gen bình thường vào tế bào gốc (stem cells) của bệnh nhân.
+ Dùng thuốc hay một phương cách nào đó để kích thích những gen tiếp tục tạo hemoglobin F. (HbF (Fetus: bào thai): tại vị trí acid amin thứ ba của chuỗi β, threonin bị thay bằng glutamic, hồng cầu biến dạng thành hình mũi tên). Loại hemoglobin này do tủy trẻ em tạo ra trong khi còn nằm trong thai nhưng thường bị "tắt" đi khi những loại hemoglobin khác bắt đầu được cấu tạo ra, do vậy nó còn được gọi là Hb bào thai.
+ Thay tủy xương.
Tài liệu tham khảo:
2. https://www.nihbt.org.vn/thalassemia/thalassemia--can-benh-nguy-hiem-can-phong-ngua/p204i21318.html
4.https://vi.wikipedia.org/wiki/B%E1%BB%87nh_tan_m%C3%A1u_b%E1%BA%A9m_sinh
5. https://hellobacsi.com/benh/thalassemia/



















