





Bệnh loạn dưỡng cơ Duchenne (DMD)
PGS.TS. Hà Hoàng Kiệm, BV 103, HVQY
Khái niệm:
Bệnh loạn dưỡng cơ tiến triển (PMD: Progressive muscular dystrophies) bao gồm các thể:
+ Loạn dưỡng cơ Duchenne - DMD (Duchenne Muscular Dystrophy) : là bệnh di truyền gen lặn liên quan đến nhiễm sắc thể X. Phụ nữ mang gen bệnh được gọi là người mang gen bệnh không có triệu chứng lâm sàng, nhưng cũng có người biểu hiện lâm sàng như yếu chi, phì đại bắp chân và nồng độ men CK tăng cao trong máu. Bệnh gặp ở trẻ em nam, có thể phát hiện bệnh khi mới sinh bằng định lượng men CK trong huyết thanh tăng cao. Các triệu chứng lâm sàng của bệnh nhi thường xuất hiện lúc 3 - 5 tuổi; Trẻ thường chậm biết đi và không chạy được một cách bình thường, trẻ có nhiều cử động nhưng tiến về phía trước thì rất khó khăn, vì trẻ không nâng đầu gối lên một cách thích hợp được; dáng đi lạch bạch và đi nhón chân xuất hiện sớm. Bệnh cơ tim giãn do loạn dưỡng cơ Duchenne (DCM: Dilated Cardiomyopathy) là thể cơ tim bị ảnh hưởng đầu tiên trong khi cơ vân thì bình thường. Những bé trai bị bệnh loạn dưỡng cơ Duchenne, do bất thường gen làm không sản sinh protein dystrophin (là một protein ở màng tế bào cơ) ở cơ của chúng. Bệnh loạn dưỡng cơ Duchenne chiếm tỉ lệ khoảng 1/3500 bé trai trên toàn thế giới.
+ Loạn dưỡng cơ Becker – BMD (Becker Muscular Dystrophy): Thể bệnh này giống loạn dưỡng cơ Duchenne ở một số đặc điểm chủ yếu sau: Liên quan đến nhiễm sắc thể X, phì đại bắp chân, yếu các cơ gốc chi là chính và nồng độ men CK huyết thanh tăng cao. Điện cơ và sinh thiết cơ giống nhau. Hai điểm khác là khởi phát thường sau 12 tuổi và tiến triển chậm hơn, còn đi được sau 20 tuổi.
+ Loạn dưỡng cơ mặt - vai - cánh tay: Bệnh di truyền theo kiểu trội, yếu cơ phân bố đặc trưng ở cơ mặt, vai, cánh tay; tiến triển chậm, khởi phát thường xảy ra ở tuổi thanh niên, ít khi ở trẻ em và nồng độ men CK huyết thanh bình thường. Đó là điểm khác nhau cơ bản với loạn dưỡng cơ Duchenne.
+ Một số thể hiếm gặp khác.
Nguyên nhân:
Bệnh xảy ra là do đột biến gen nằm trên nhiễm sắc thể giới tính X (được phát hiện năm 1986) chịu trách nhiệm sản xuất dystropin (phát hiện năm 1987). Gen chịu trách nhiệm sản xuất dystropin là gen lớn nhất con người từng phát hiện (2.3 megabases/Xp21.2), trọng lượng protein của nó là 427 kD. Gen biến dị dạng thiếu hụt exon xảy ra trong 60 - 65% bệnh Những bé trai bị bệnh loạn dưỡng cơ Duchenne, do bất thường gen làm không sản sinh protein dystrophin (là một protein ở màng tế bào cơ) ở cơ của chúng. Bệnh loạn dưỡng cơ Duchenne chiếm tỉ lệ khoảng 1/3500 bé trai trên toàn thế giới. 65 - 70% bệnh loạn dưỡng cơ Becker bị lặp đoạn 1 phần trong 5 - 10% trường hợp, còn lại là do thiếu hụt nucleotide đơn độc và nhiều thiếu hụt khác trong quá trình giải mã. Một số trường hợp biểu hiện bệnh loạn dưỡng cơ Duchenne hoặc bệnh loạn dưỡng cơ Becker trên lâm sàng nhưng không di truyền từ nhiễm sắc thể X có lẽ do thiếu hụt các gen mã hóa cho glycoprotein đi kèm với dystropin.
Trong đa số trường hợp, bệnh loạn dưỡng cơ Duchenne là do gián đoạn mạng lưới giải mã dystropin trong khi loạn dưỡng cơ Becker thì gồm các đột biến vẫn bảo tồn chuỗi amino acid. Protein dystropin nằm ở mặt tương bào của màng bào tương của tế bào cơ. Dystropin cung cấp sự vững chắc cơ học cho màng tế bào cơ (sarcolemma = màng tế bào bao bọc tế bào cơ) và ổn định phức hợp glycoprotein nhằm ngăn thoái biến tế bào sợi cơ do protease. Dystropin đảm bảo cho hoạt động của các protein đi chung với nó như nNOS, cần thiết cho sự sản xuất NO (có tác dụng giãn mạch và tăng dòng máu đến cơ) giúp ngăn mỏi cơ khi vận động gắng sức. Màng tế bào cơ bị tổn thương do thiếu dystropin khiến dòng calci ngoại bào đổ vào tế bào cơ. Các yếu tố điều hòa viêm sinh ra từ cơ bị loạn dưỡng làm tăng iNOS, đưa đến làm mất ổn định các receptor ryanodine trên lưới nội bào điều hòa dòng calci đi vào. Nồng độ calci trong bào tương tăng cao, hoạt hóa calpain thúc đẩy ly giải cơ.
Các biến thể của gen LTBP4 liên quan đến tuổi biểu hiện bệnh loạn dưỡng cơ Duchenne. Các biến thể của SPP1 liên quan đến giảm sức cơ và mất vận động ở tuổi trẻ hơn của bệnh loạn dưỡng cơ Duchenne, dng5 này đáp ứng với điều trị bằng glucocorticoid.
Bệnh chỉ gặp ở bệnh nhi nam. Di truyền gen từ mẹ sang con trai theo kiểu di truyền lặn. Gen khiếm khuyết nằm trên một nhiễm sắc thể giới tính X. Người phụ nữ mang gen bệnh trên nhiễm sắc thể X nhưng chỉ là người mang gen và không biểu hiện bệnh do nhiễm sắc thể X còn lại bình thường. Khi thụ tinh, noãn mang nhiễm sắc thể X có gen bệnh kết hợp với tinh trùng mang nhiễm sắc thể Y và bé trai con của người được thừa hưởng gen này trên nhiễm sắc thể X, có biểu hiện bệnh. Trong một số trường hợp bệnh phát sinh từ một đột biến mới ở gen khác chứ không phải là từ gen khiếm khuyết di truyền.
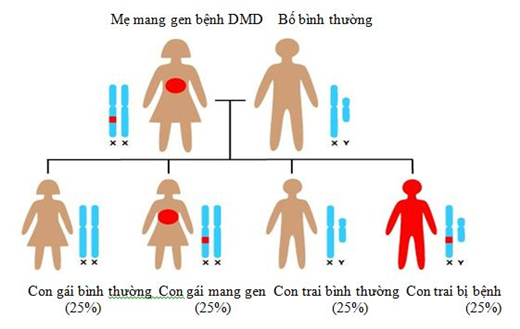
Hình 1. Sơ đồ di truyền gen bệnh loạn dưỡng cơ Duchenne từ mẹ sang con.
Biểu hiện lâm sàng:
Bệnh biểu hiện thoái hóa cơ vân tiến triển, không có tổn thương thần kinh.
- Tuổi khởi phát: thường 2 - 3 tuổi, gặp ở trẻ nam, một số trường hợp biểu hiện muộn hơn.
- Có thể kèm ảnh hưởng phát triển trí tuệ hoặc không.
- Yếu cơ: Ðầu tiên bệnh ảnh hưởng tới các cơ vùng chậu, cánh tay và đùi. Loạn dưỡng cơ Duchenne thường xuất hiện ở các bé trai và là dạng loạn dưỡng cơ phổ biến nhất ở trẻ em. Yếu cơ gốc chi nặng hơn ngọn chi, chi dưới nặng hơn chi trên nên khó đi lại, nhảy hay chạy.
- Dấu hiệu Gower: Trẻ dùng tay chống đẩy người để ngồi thẳng dậy; đi lạch bạch, vẹo cột sống hoặc đùi to là dấu hiệu thường gặp kèm đau hoặc không.
- Bệnh cơ tim: Bệnh loạn dưỡng cơ Duchenne gây ra bệnh cơ tim giãn và các dạng loạn nhịp chủ yếu ở thất, đặc trưng bởi sự xơ hóa quá mức vùng sau dưới thất trái đưa đến những thay đổi điện tim như R cao ở các chuyển đạo trước ngực kèm tăng tỉ số R/S, Q sâu ở DI, aVL, V5 - V6. Khi tiến triển, xơ hóa lan đến thành bên thất trái, gây hở van 2 lá do ảnh hưởng cơ nhú sau. Tỷ lệ mắc mới tăng dần từ 1/3 năm 14 tuổi, lên 1/2 năm 18 tuổi và 100% khi trên 18 tuổi. Mặc dù tỷ lệ mới mắc bệnh cơ tim giãn cao nhưng đa số lại được phát hiện muộn. Đôi khi, gây đột tử mà không có bất thường về hô hấp nào.
- Biến chứng cơ xương khớp: gãy xương do ngã, vẹo cột sống và yếu cơ tiến triển gây ảnh hưởng lên hô hấp đưa đến suy hô hấp. Khám: giả phì đại cơ đùi và cơ tứ đầu đùi, đi lạch bạch, ngắn gót chân, vẹo cột sống, giảm hoặc mất phản xạ.
- Loạn dưỡng cơ Becker: tuổi khởi phát muộn hơn loạn dưỡng cơ Duchenne với mức độ nhẹ hơn, thường khoảng 15 tuổi và có thể sống khỏe đến năm trưởng thành, vài bệnh nhân duy trì được vận động đến già; có sự bảo tồn tương đối sức cơ gập cổ. Phân biệt giữa loạn dưỡng cơ Becker và loạn dưỡng cơ chi - vai thường khó khi không có tiền sử gia đình nhưng loạn dưỡng cơ chi - vai thường không có giả phì đại cơ đùi.
Bệnh loạn dưỡng cơ Becker thường ảnh hưởng đến cơ tim rõ ràng hơn so với bệnh loạn dưỡng cơ Duchenne, do thể loạn dưỡng cơ Becker bệnh nhân vẫn có thể vận động gắng sức đã tạo áp lực lên cơ tim có rối loạn dystropin. Thất phải bị ảnh hưởng sớm hơn thất trái, khi xơ hóa tiến triển đưa đến suy tim tiến triển nặng. Bệnh cơ tim giãn do loạn dưỡng cơ Duchenne biểu hiện suy tim sung huyết xuất hiện từ 20 – 40 tuổi, tiến triển nhanh đến tử vong.
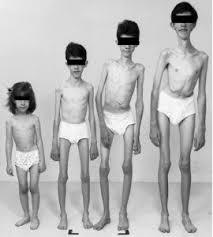
Hình 2. Bệnh nhân bị bệnh loạn dưỡng cơ Duchenne.
Teo cơ gốc chi đối xứng (lưng, ngực, vai...) và bắp chân phì đại, giai đoạn cuối trẻ mất khả năng đi lại, suy tim và suy hô hấp. Co rút gân Achille làm khi bước trẻ có tư thế kiễng chân. Mất cơ lực tăng dần chủ yếu ở các gốc chi và các cơ gấp của cổ làm cho lệch trục chi và cổ. Tổn thương cơ ở chi dưới thường nặng hơn chi trên. Nhiều bệnh nhân không rời khỏi được xe lăn, co cứng cơ làm gù lưng tăng dần, biến dạng lồng ngực kèm gù lưng làm cho chức năng hô hấp vốn đã giảm lại càng giảm vì yếu cơ. Phản xạ gân xương giảm, không có phản xạ bệnh lý bó tháp, không có rối loạn cảm giác.
Cận lâm sàng:
- Creatinine kinase (CK): tăng gấp 10 - 20 lần so với giới hạn trên của bình thường, sau đó giảm dần về bình thường sau khi cơ được thay thế bởi mô sợi và mỡ. CK tăng 3 lần trở lên so với giới hạn trên bình thường ở 50 - 70% bệnh nhân.
- Điện cơ: biểu hiện bệnh cơ, điện thế đa pha nhỏ. Điện thần kinh: bình thường.
- Sinh thiết cơ: thoái hóa, tái sinh và giả phì đại sợi cơ, thay thế bằng mô mỡ và mô liên kết.
- Phân tích dystropin: ở người bình thường, dystropin có thể thăm dò bằng màng thấm miễn dịch chứa 100μg protein cơ, sau đó được xác định bằng mắt hay bằng phương pháp đo tỷ trọng: Gần 100% bệnh nhân loạn dưỡng cơ Duchenne không có dystropin. Khoảng 85% loạn dưỡng cơ Becker có bất thường phân tử dystropin.
- Miễn dịch thấm màng dystropin có thể dùng để định độ nặng của phân típ loạn dưỡng cơ, số lượng dystropin quyết định độ nặng của loạn dưỡng cơ Duchenne trong khi chất lượng dystropin quyết định độ nặng của loạn dưỡng cơ Becker: bệnh loạn dưỡng cơ Duchenne chỉ có dưới 5% số lượng dystropin có kích thước phân tử bình thường; 5 - 20% số lượng dystropin có kích thước phân tử bình thường có liên quan với các phân típ trung gian. 20 - 50% số lượng dystropin kích thước bình thường hoặc 20 - 100% số lượng dystropin có kích thước bình thường đi kèm với loạn dưỡng cơ Becker nhẹ đến trung bình.
Chẩn đoán:
Nghi ngờ bệnh loạn dưỡng cơ khi có các dấu hiệu sau:
- Giới tính nam và tuổi 2 - 3.
- Sự xuất hiện triệu trứng phản ánh diễn tiến bệnh.
- Tăng CK huyết tương.
- Những thay đổi trên điện cơ và sinh thiết cơ, trong khi điện thần kinh bình thường.
- Tiền sử gia đình di truyền liên quan đến nhiễm sắc thể X.
- Phân tích gen thấy thiếu đoạn/lặp đoạn (70 - 80% trường hợp) hoặc đột biến điểm (20 - 30% trường hợp): MLPA (Multiplex ligation dependent probe amplification) giúp thăm dò thiếu/lặp đoạn. Sàng lọc bằng DHPLC, phương pháp chuỗi trực tiếp, phương pháp SSCP, DOVAM-S, SCAIP, DGGE sàng lọc đột biến điểm… Sự xuất hiện của LGMD2l như DMD/BMD: do biến dị gen FKRP, một số phân típ bệnh nhân của bệnh DMD/BMD có test dystropin âm tính nên kiểm tra gen FKRP.
- Sinh thiết cơ cần thiết trong các trường hợp sau:
+ Trường hợp lâm sàng không điển hình.
+ Gia đình không có bệnh cảnh di truyền nhiễm sắc thể X.
+ Gia đình có anh/chị em cùng bị gợi ý dạng thoái triển tự thân của bệnh cơ loạn dưỡng.
Tuy nhiên gần đây, vai trò sinh thiết cơ trong chẩn đoán được thay thế bởi các xét nghiệm về gen.
Điều trị:
Chưa có biện pháp điều trị khỏi. Điều trị bệnh di truyền loạn dưỡng cơ Duchenne là điều trị triệu chứng. Theo dõi tích cực bệnh cơ tim giãn, sử dụng thuốc làm giảm ứ huyết tim, bao gồm cả ghép tim trong trường hợp nặng. Có thể cần các thiết bị trợ giúp hô hấp khi có biến chứng hô hấp, đặc biệt là vào ban đêm. Thuốc prednisone được sử dụng để cải thiện sức mạnh cơ và chức năng của các bệnh nhân loạn dưỡng cơ Duchenne. Prednisone đã được chứng minh có thể kéo dài khả năng đi lại từ 2 đến 5 năm. Tuy nhiên, tác dụng phụ của prednisone bao gồm tăng cân, huyết áp cao, thay đổi hành vi và tăng trưởng chậm. Thuốc Emflaza (deflazacort) là một corticosteroid vừa được Cơ quan Quản lý Dược phẩm và Thực phẩm Mỹ (FDA) phê duyệt để điều trị cho bệnh nhân từ 5 tuổi trở lên bị bệnh teo cơ Duchenne ít tác dụng phụ hơn so với prednisolon. Thuốc ức chế miễn dịch cyclosporin đã được thử nghiệm tỏ ra cải thiện được chức năng lâm sàng ở trẻ em, nhưng việc sử dụng thuốc này gây nhiều tranh cãi do cyclosporin có thể gây ra bệnh cơ. Oxandrolone, một loại thuốc được sử dụng trong một nghiên cứu, có tác dụng tương tự như prednisone với ít tác dụng phụ. Một số phương pháp điều trị khác cũng đang được nghiên cứu, bao gồm coenzyme Q10, glutamine, pentoxifylline và PTC124.
Vật lý trị liệu được sử dụng để thúc đẩy khả năng vận động và phòng ngừa co rút cơ. Phẫu thuật có thể cần thiết khi có co cứng nghiêm trọng và vẹo cột sống. Vận động trị liệu, hoạt động trị liệu và điện trị liệu thường được áp dụng.
Tiên lượng và phòng bệnh:
Đa số bệnh nhi tử vong vào cuối tuổi vị thành niên hoặc ngoài 20 tuổi, thường do viêm phổi, yếu cơ hô hấp hoặc các biến chứng tim. Một số người bị loạn dưỡng cơ Duchenne có biểu hiện vẹo cột sống.
Bệnh loạn dưỡng cơ Duchenne được di truyền lặn liên kết với nhiễm sắc thể giới tính X. Nam giới chỉ nhận một bản sao nhiễm sắc thể X từ mẹ và bản sao nhiễm sắc thể Y từ cha. Nếu nhiễm sắc thể X của họ có một đột biến gen loạn dưỡng cơ Duchenne, họ sẽ mắc bệnh loạn dưỡng cơ Duchenne. Mặt khác, ở phụ nữ do có hai nhiễm sắc thể X, nếu một bản sao không làm việc, người nữ có bản sao thứ hai để sản xuất các protein dystrophin. Một phụ nữ có biến đổi di truyền ở một trong hai bản sao của họ được cho là “người mang gen” bệnh loạn dưỡng cơ Duchenne. Người mang gen không biểu hiện bệnh loạn dưỡng cơ Duchenne và hầu hết họ không biết mình có đột biến trong vật chất di truyền trừ khi họ có tiền sử bệnh trong gia đình. Tuy nhiên, nghiên cứu gần đây đã chỉ ra rằng một số phụ nữ mang gen bệnh (khoảng 20 phần trăm) sẽ có triệu chứng của bệnh loạn dưỡng cơ Duchenne, bao gồm yếu cơ và bệnh tim giãn. Với sự di truyền lặn liên kết với X, gen đột biến (không làm việc) trên bản sao nhiễm sắc thể của một đứa trẻ là khác nhau ở nam và nữ. Những người phụ nữ mang các bản sao đột biến gen sẽ truyền cho 50% con khi mai thai. Do đó, 25% con có biểu hiện bệnh loạn dưỡng cơ Duchenne (ví dụ, 50% bé trai có xác suất mắc bệnh loạn dưỡng cơ Duchenne và 50% các bé gái sẽ là những người mang gen bệnh). Xác suất một người phụ nữ đã có một con trai bị bệnh (và không có tiền sử gia đình) mang gen bệnh loạn dưỡng cơ Duchenne là khoảng 2/3. Tuy nhiên, ở một phần ba còn lại của các cá nhân loạn dưỡng cơ Duchenne, sự thay đổi trong gen dystrophin là một sự thay đổi di truyền mới và khoảng 10% đột biến mới này là do khảm tuyến sinh dục. Khảm sinh dục nghĩa là một cá nhân có hai hay nhiều quần thể tế bào khác nhau trong cấu trúc di truyền trong trứng hoặc tinh trùng của họ. Nam giới thừa kế hoặc được sinh ra với một bản sao đột biến của gen loạn dưỡng cơ Duchenne sẽ mắc bệnh loạn dưỡng cơ Duchenne vì họ có một nhiễm sắc thể Y và không có nhiễm sắc thể X thứ hai. Nếu một người đàn ông với gen bệnh loạn dưỡng cơ Duchenne có con, tất cả các con gái của ông sẽ là thể mang gen và không ai trong số con trai của ông sẽ bị bệnh.
Khoa học có thể cho phép lựa chọn sinh sản khác nhau cho các gia đình. Đã có công nghệ có thể tách tinh trùng chứa nhiễm sắc thể X mang gen bệnh. Lựa chọn sinh sản thứ hai là chẩn đoán di truyền (hay xét nghiệm ADN), đó là kỹ thuật cho phép các tế bào của trứng đã thụ tinh được thử nghiệm để xác định xem nó có hay không chứa đột biến trong gen loạn dưỡng cơ Duchenne và sau đó cấy những quả trứng nào không mang gen bệnh. Các xét nghiệm có thể lựa chọn: Lấy mẫu lông nhung màng đệm (CVS) và chọc ối, phân tích các tế bào lấy mẫu có nguồn gốc từ phôi đang phát triển.
Một số lựa chọn xét nghiệm tiền sản cho thai khi nguy cơ có bệnh loạn dưỡng cơ Duchenne đã được xác định trong một thành viên gia đình, hoặc nếu thông tin đánh dấu di truyền liên kết đã được xác định bởi xét nghiệm AND.
Tài liệu tham khảo:
https://www.slideshare.net/longlexuan4/bnh-duchenne-lm-sng-v-chn-on
http://xetnghiemdna.com/2013/10/benh-loan-duong-co-la-gi/
https://www.voatiengviet.com/a/hoi-dap-y-hoc-benh-co-loan-duong-duchenne/2547764.html



















